CNRS UMR 6061, Institut de Génétique et Développement de Rennes, IFR 140 GFAS, Faculté de Médecine, 2 avenue du Professeur Léon Bernard CS 34317, 35043 Rennes Cedex, France.
Orphanet J Rare Dis. 2011 Mar 15;6:9. doi: 10.1186/1750-1172-6-9.
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is characterized by congenital aplasia of the uterus and the upper part of the vagina in women showing normal development of secondary sexual characteristics and a normal 46, XX karyotype. The uterovaginal aplasia is either isolated (type I) or more frequently associated with other malformations (type II or Müllerian Renal Cervico-thoracic Somite (MURCS) association), some of which belong to the malformation spectrum of DiGeorge phenotype (DGS). Its etiology remains poorly understood. Thus the phenotypic manifestations of MRKH and DGS overlap suggesting a possible genetic link. This would potentially have clinical consequences.
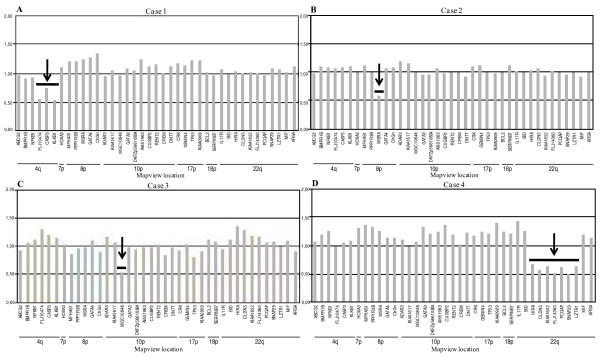
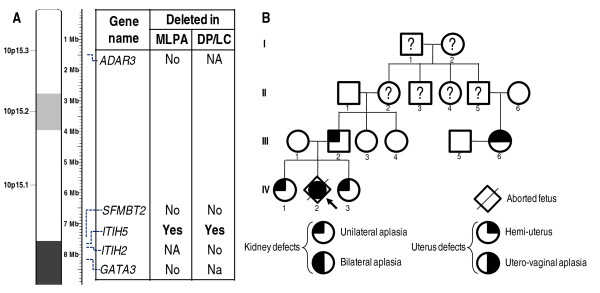
We searched DiGeorge critical chromosomal regions for chromosomal anomalies in a cohort of 57 subjects with uterovaginal aplasia (55 women and 2 aborted fetuses). For this candidate locus approach, we used a multiplex ligation-dependent probe amplification (MLPA) assay based on a kit designed for investigation of the chromosomal regions known to be involved in DGS.The deletions detected were validated by Duplex PCR/liquid chromatography (DP/LC) and/or array-CGH analysis.
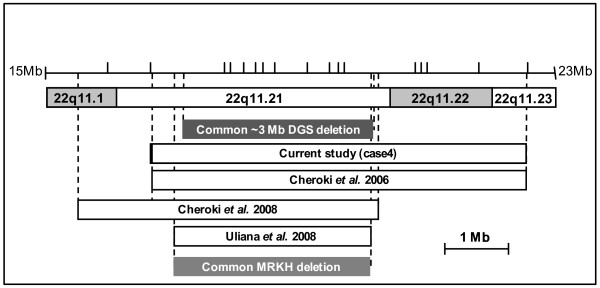
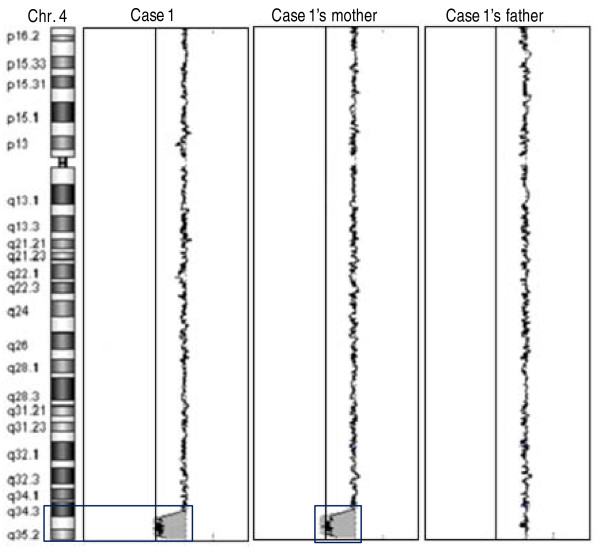
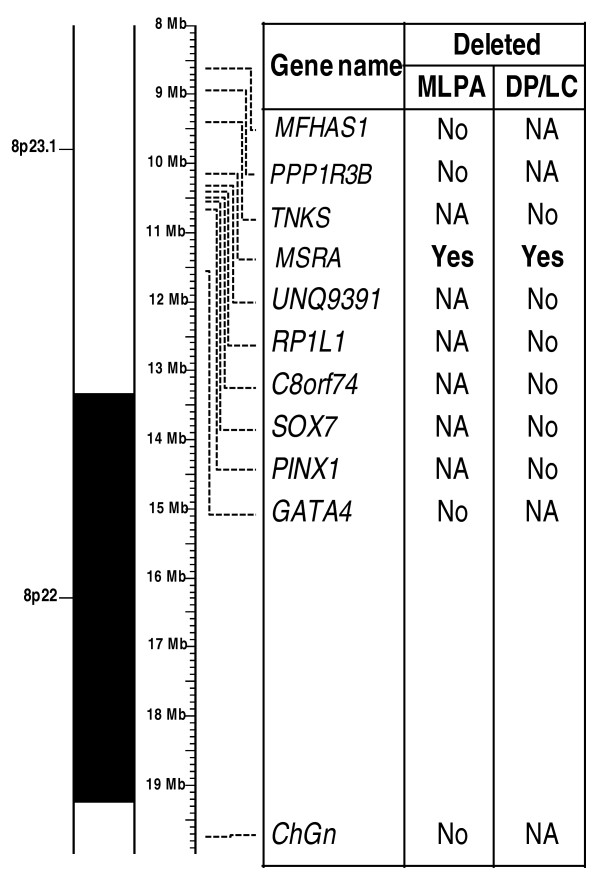
We found deletions in four probands within the four chromosomal loci 4q34-qter, 8p23.1, 10p14 and 22q11.2 implicated in almost all cases of DGS syndrome.
Uterovaginal aplasia appears to be an additional feature of the broad spectrum of the DGS phenotype. The DiGeorge critical chromosomal regions may be candidate loci for a subset of MRKH syndrome (MURCS association) individuals. However, the genes mapping at the sites of these deletions involved in uterovaginal anomalies remain to be determined. These findings have consequences for clinical investigations, the care of patients and their relatives, and genetic counseling.
Mayer-Rokitansky-Küster-Hauser (MRKH) 综合征的特征是女性子宫和阴道上部先天性发育不全,表现为第二性征正常发育和正常的 46,XX 核型。子宫阴道发育不全要么是孤立的(I 型),要么更常与其他畸形相关(II 型或 Müllerian Renal Cervico-thoracic Somite (MURCS) 关联),其中一些属于 DiGeorge 表型(DGS)的畸形谱。其病因仍知之甚少。因此,MRKH 和 DGS 的表型表现重叠,表明可能存在遗传联系。这可能会产生临床后果。
我们在一个子宫阴道发育不全的 57 名受试者队列中(55 名女性和 2 名流产胎儿),对 DiGeorge 关键染色体区域进行了染色体异常搜索。对于这种候选基因座方法,我们使用了一种基于针对涉及 DGS 的染色体区域设计的试剂盒的多重连接依赖性探针扩增(MLPA)检测。检测到的缺失通过 Duplex PCR/液相色谱(DP/LC)和/或阵列-CGH 分析进行验证。
我们在四个染色体基因座 4q34-qter、8p23.1、10p14 和 22q11.2 中的四个先证者中发现了缺失,这些缺失几乎涉及所有 DGS 综合征病例。
子宫阴道发育不全似乎是 DGS 表型广泛谱的另一个特征。DiGeorge 关键染色体区域可能是一部分 MRKH 综合征(MURCS 关联)个体的候选基因座。然而,映射到涉及子宫阴道异常的这些缺失部位的基因仍有待确定。这些发现对临床研究、患者及其亲属的护理以及遗传咨询具有影响。