Department of Clinical Genetics, Aalborg University Hospital, Aalborg, Denmark.
Department of Clinical Genetics, Aarhus University Hospital, Brendstrupgårdsvej 21C, DK-8200, Aarhus N, Denmark.
Orphanet J Rare Dis. 2020 Aug 20;15(1):214. doi: 10.1186/s13023-020-01491-9.
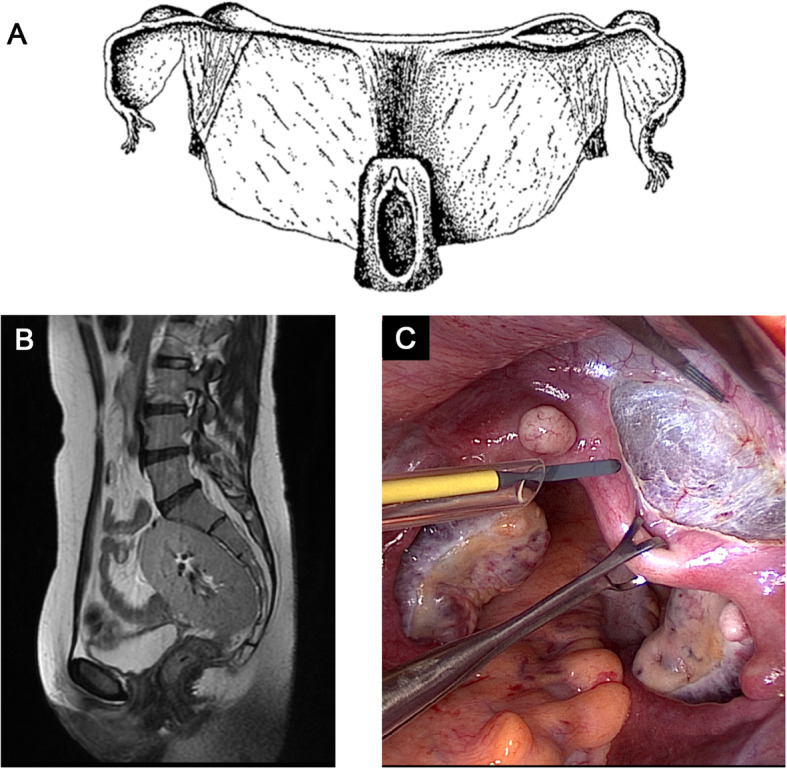
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, also referred to as Müllerian aplasia, is a congenital disorder characterized by aplasia of the uterus and upper part of the vagina in females with normal secondary sex characteristics and a normal female karyotype (46,XX).
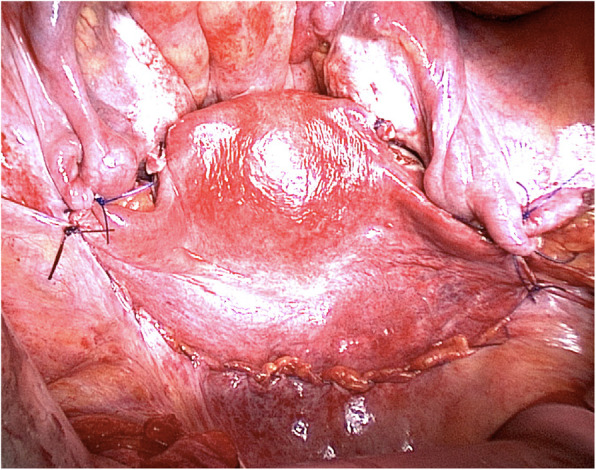
The diagnosis is often made during adolescence following investigations for primary amenorrhea and has an estimated prevalence of 1 in 5000 live female births. MRKH syndrome is classified as type I (isolated uterovaginal aplasia) or type II (associated with extragenital manifestations). Extragenital anomalies typically include renal, skeletal, ear, or cardiac malformations. The etiology of MRKH syndrome still remains elusive, however increasing reports of familial clustering point towards genetic causes and the use of various genomic techniques has allowed the identification of promising recurrent genetic abnormalities in some patients. The psychosexual impact of having MRKH syndrome should not be underestimated and the clinical care foremost involves thorough counselling and support in careful dialogue with the patient. Vaginal agenesis therapy is available for mature patients following therapeutical counselling and education with non-invasive vaginal dilations recommended as first-line therapy or by surgery. MRKH syndrome involves absolute uterine factor infertility and until recently, the only option for the patients to achieve biological motherhood was through gestational surrogacy, which is prohibited in most countries. However, the successful clinical trial of uterus transplantation (UTx) by a Swedish team followed by the first live-birth in September, 2014 in Gothenburg, proofed the first available fertility treatment in MRKH syndrome and UTx is now being performed in other countries around the world allowing women with MRKH syndrome to carry their own child and achieve biological motherhood.
Several advances in research across multiple disciplines have been made in the recent years and this kaleidoscopic review provides a current status of various key aspects in MRKH syndrome and provides perspectives for future research and improved clinical care.
Mayer-Rokitansky-Küster-Hauser(MRKH)综合征,也称为 Müllerian 发育不全,是一种先天性疾病,其特征为女性的子宫和阴道上段发育不全,具有正常的第二性征和正常的女性核型(46,XX)。
该疾病通常在青春期因原发性闭经而进行检查时被诊断,其预估患病率为每 5000 例活产女婴中有 1 例。MRKH 综合征分为 I 型(孤立性子宫阴道发育不全)或 II 型(伴有外生殖器表现)。外生殖器异常通常包括肾脏、骨骼、耳朵或心脏畸形。MRKH 综合征的病因仍不清楚,然而,越来越多的家族聚集报告表明其与遗传有关,并且各种基因组技术的应用使得一些患者中潜在的复发性遗传异常得以识别。MRKH 综合征对患者的心理影响不应被低估,临床护理首先包括与患者进行彻底的咨询和支持,进行仔细的对话。在经过治疗咨询和教育后,为成熟患者提供阴道发育不全的治疗,推荐非侵入性阴道扩张作为一线治疗或手术治疗。MRKH 综合征涉及绝对子宫因素不孕,直到最近,患者实现生物学母亲身份的唯一选择是通过代孕,而这在大多数国家都是被禁止的。然而,瑞典团队成功进行了子宫移植(UTx)的临床试验,随后于 2014 年 9 月在哥德堡进行了首例活产,证明了 MRKH 综合征中第一种可行的生育治疗方法,UTx 现已在世界其他国家进行,允许 MRKH 综合征患者拥有自己的孩子并实现生物学母亲身份。
近年来,在多个学科领域的研究取得了多项进展,本综述提供了 MRKH 综合征的各个关键方面的最新状况,并为未来的研究和改善临床护理提供了视角。