Protein Metabolism Project, Tokyo Metropolitan Institute of Medical Science, Setagaya-ku, Tokyo 156-8501, Japan.
J Cell Biol. 2011 Apr 18;193(2):275-84. doi: 10.1083/jcb.201102031. Epub 2011 Apr 11.
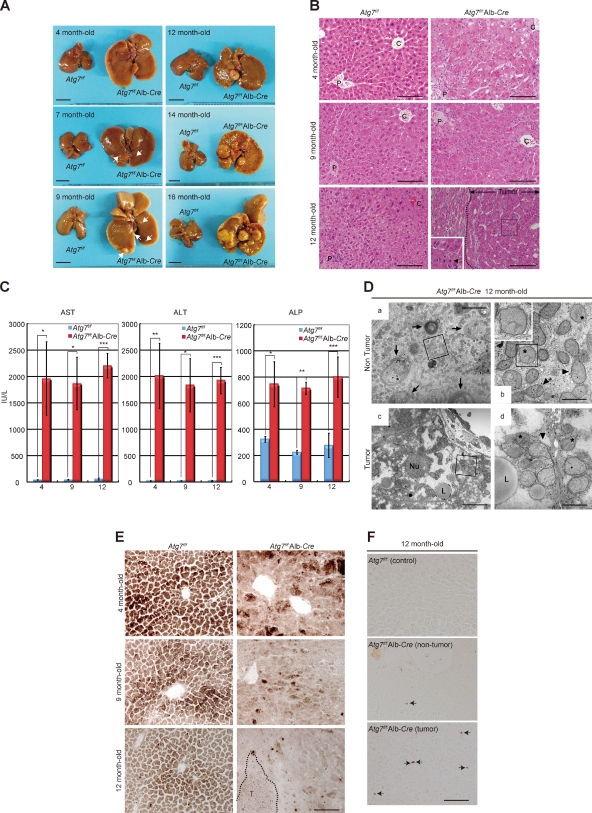
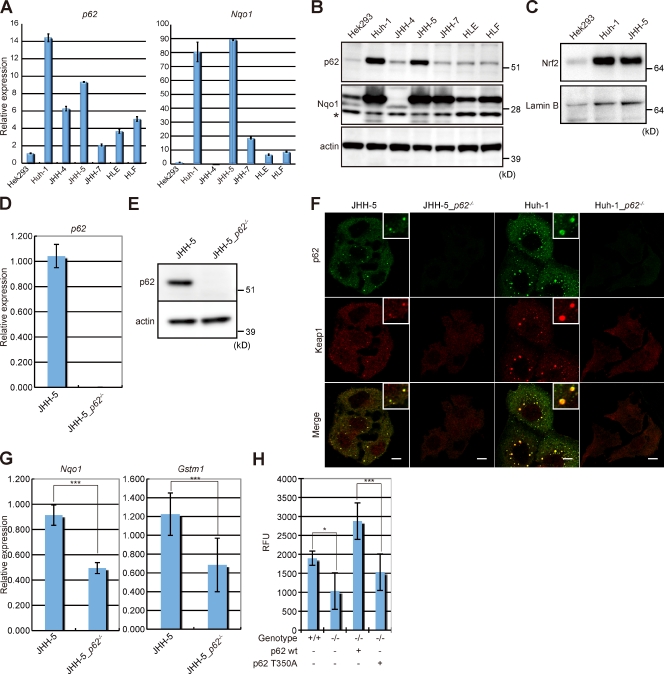
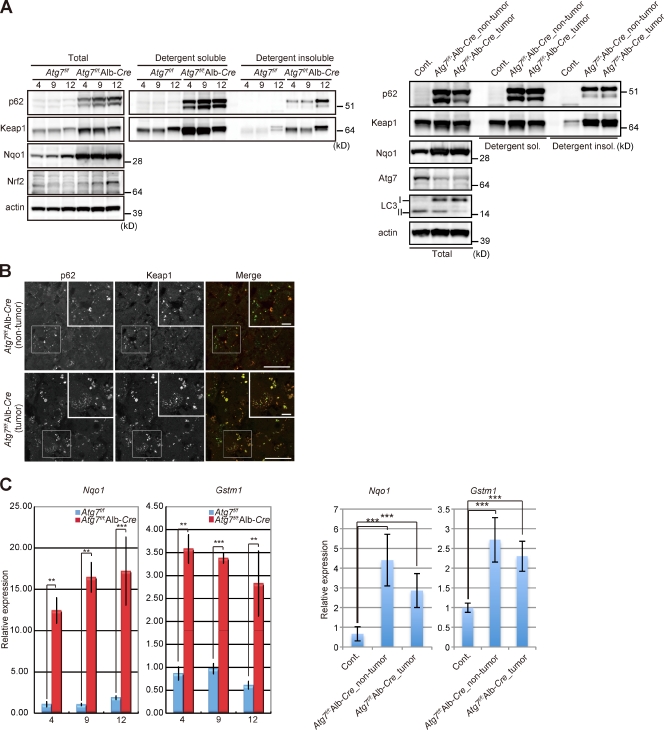
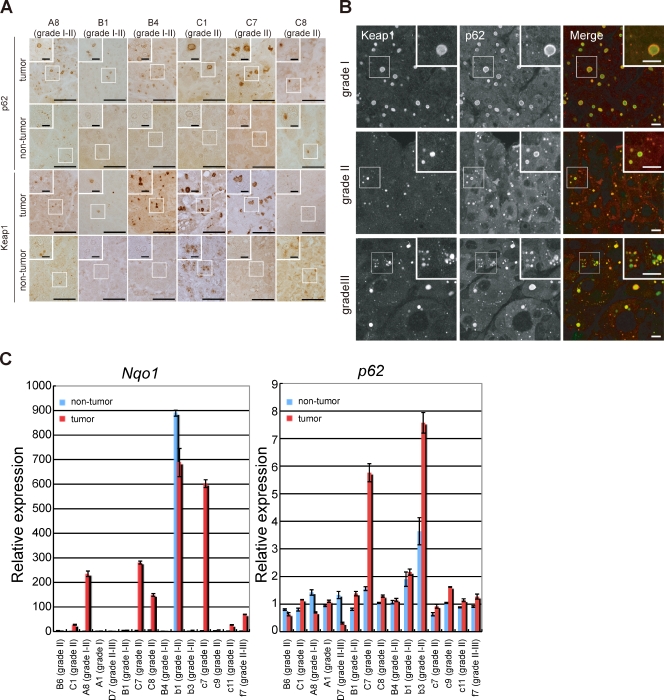
Suppression of autophagy is always accompanied by marked accumulation of p62, a selective autophagy substrate. Because p62 interacts with the Nrf2-binding site on Keap1, which is a Cullin 3-based ubiquitin ligase adapter protein, autophagy deficiency causes competitive inhibition of the Nrf2-Keap1 interaction, resulting in stabilization of Nrf2 followed by transcriptional activation of Nrf2 target genes. Herein, we show that liver-specific autophagy-deficient mice harbor adenomas linked to both the formation of p62- and Keap1-positive cellular aggregates and induction of Nrf2 targets. Importantly, similar aggregates were identified in more than 25% of human hepatocellular carcinomas (HCC), and induction of Nrf2 target genes was recognized in most of these tumors. Gene targeting of p62 in an HCC cell line markedly abrogates the anchorage-independent growth, whereas forced expression of p62, but not a Keap1 interaction-defective mutant, resulted in recovery of the growth defect. These results indicate the involvement of persistent activation of Nrf2 through the accumulation of p62 in hepatoma development.
自噬的抑制总是伴随着 p62 的明显积累,p62 是一种选择性自噬底物。由于 p62 与 Keap1 上的 Nrf2 结合位点相互作用,而 Keap1 是一种基于 Cullin3 的泛素连接酶衔接蛋白,因此自噬缺陷导致 Nrf2-Keap1 相互作用的竞争性抑制,导致 Nrf2 稳定化,随后 Nrf2 靶基因转录激活。在此,我们表明,肝脏特异性自噬缺陷小鼠存在与 p62 和 Keap1 阳性细胞聚集形成以及 Nrf2 靶基因诱导相关的腺瘤。重要的是,在超过 25%的人类肝细胞癌 (HCC) 中鉴定到了类似的聚集物,并且在这些肿瘤中的大多数中都识别到了 Nrf2 靶基因的诱导。在 HCC 细胞系中靶向 p62 的基因敲除显著阻断了锚定非依赖性生长,而强制表达 p62(而非 Keap1 相互作用缺陷突变体)则导致生长缺陷得到恢复。这些结果表明,通过 p62 的积累,持续激活 Nrf2 参与了肝癌的发生。