Lee Se Eun, Jung Yun Hye, Han Kyoung Hee, Lee Hyun Kyung, Kang Hee Gyung, Ha Il Soo, Choi Yong, Cheong Hae Il
Department of Pediatrics, Seoul National University Children's Hospital, Seoul, Korea.
Korean J Pediatr. 2011 Feb;54(2):90-3. doi: 10.3345/kjp.2011.54.2.90. Epub 2011 Feb 28.
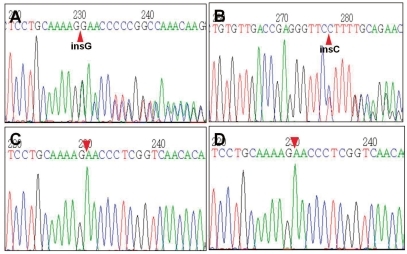
Pseudohypoaldosteronism type 1 (PHA1) is a rare form of mineralocorticoid resistance characterized in newborns by salt wasting with dehydration, hyperkalemia and failure to thrive. This disease is heterogeneous in etiology and includes autosomal dominant PHA1 owing to mutations of the NR3C2 gene encoding the mineralocorticoid receptor, autosomal recessive PHA1 due to mutations of the epithelial sodium channel (ENaC) gene, and secondary PHA1 associated with urinary tract diseases. Amongst these diseases, autosomal dominant PHA1 shows has manifestations restricted to renal tubules including a mild salt loss during infancy and that shows a gradual improvement with advancing age. Here, we report a neonatal case of PHA1 with a NR3C2 gene mutation (a heterozygous c.2146_2147insG in exon 5), in which the patient showed failure to thrive, hyponatremia, hyperkalemia, and elevated plasma renin and aldosterone levels. This is the first case of pseudohypoaldosteronism type 1 confirmed by genetic analysis in Korea.
1型假性醛固酮增多症(PHA1)是一种罕见的盐皮质激素抵抗形式,在新生儿中表现为失盐伴脱水、高钾血症和生长发育迟缓。该病病因具有异质性,包括由于编码盐皮质激素受体的NR3C2基因突变导致的常染色体显性PHA1、由于上皮钠通道(ENaC)基因突变导致的常染色体隐性PHA1以及与泌尿系统疾病相关的继发性PHA1。在这些疾病中,常染色体显性PHA1的表现局限于肾小管,包括婴儿期轻度失盐,且随着年龄增长逐渐改善。在此,我们报告1例携带NR3C2基因突变(外显子5杂合c.2146_2147insG)的新生儿PHA1病例,该患者表现为生长发育迟缓、低钠血症、高钾血症以及血浆肾素和醛固酮水平升高。这是韩国首例经基因分析确诊的1型假性醛固酮增多症病例。