Inserm U692, Laboratoire de Signalisations Moléculaires et Neurodégénérescence, Strasbourg, F-67085 France.
Mol Neurodegener. 2011 Apr 26;6(1):26. doi: 10.1186/1750-1326-6-26.
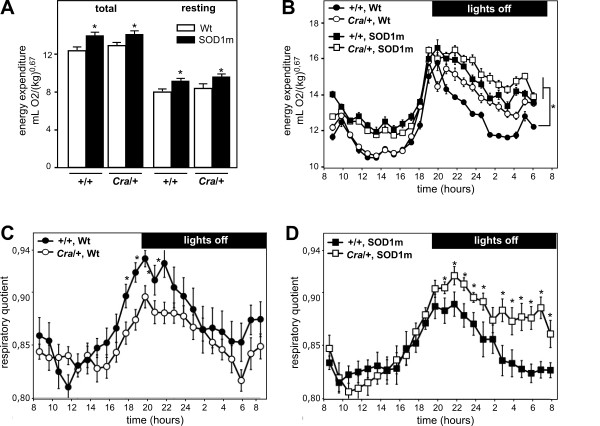
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by a progressive loss of motor neurons. ALS patients, as well as animal models such as mice overexpressing mutant SOD1s, are characterized by increased energy expenditure. In mice, this hypermetabolism leads to energy deficit and precipitates motor neuron degeneration. Recent studies have shown that mutations in the gene encoding the dynein heavy chain protein are able to extend lifespan of mutant SOD1 mice. It remains unknown whether the protection offered by these dynein mutations relies on a compensation of energy metabolism defects.
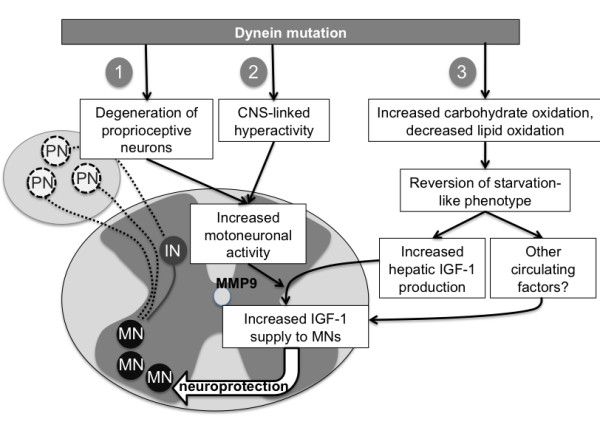
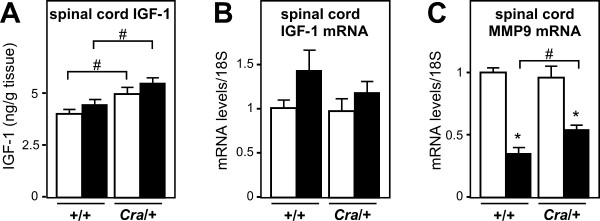
SOD1(G93A) mice were crossbred with mice harboring the dynein mutant Cramping allele (Cra/+ mice). Dynein mutation increased adipose stores in compound transgenic mice through increasing carbohydrate oxidation and sparing lipids. Metabolic changes that occurred in double transgenic mice were accompanied by the normalization of the expression of key mRNAs in the white adipose tissue and liver. Furthermore, Dynein Cra mutation rescued decreased post-prandial plasma triglycerides and decreased non esterified fatty acids upon fasting. In SOD1(G93A) mice, the dynein Cra mutation led to increased expression of IGF-1 in the liver, increased systemic IGF-1 and, most importantly, to increased spinal IGF-1 levels that are potentially neuroprotective.
These findings suggest that the protection against SOD1(G93A) offered by the Cramping mutation in the dynein gene is, at least partially, mediated by a reversal in energy deficit and increased IGF-1 availability to motor neurons.
肌萎缩侧索硬化症(ALS)是一种致命的神经退行性疾病,其特征是运动神经元逐渐丧失。ALS 患者以及过度表达突变 SOD1 的小鼠等动物模型的特点是能量消耗增加。在小鼠中,这种代谢亢进导致能量不足并促使运动神经元退化。最近的研究表明,编码动力蛋白重链蛋白的基因突变能够延长突变 SOD1 小鼠的寿命。尚不清楚这些动力蛋白突变提供的保护是否依赖于对能量代谢缺陷的补偿。
SOD1(G93A)小鼠与携带动力蛋白突变 Cramping 等位基因(Cra/+ 小鼠)的小鼠进行杂交。动力蛋白突变通过增加碳水化合物氧化和节省脂质来增加复合转基因小鼠的脂肪储存。双转基因小鼠发生的代谢变化伴随着白色脂肪组织和肝脏中关键 mRNA 表达的正常化。此外,Dynein Cra 突变可纠正 SOD1(G93A)小鼠进食后血浆甘油三酯和空腹时非酯化脂肪酸减少的现象。在 SOD1(G93A)小鼠中,动力蛋白 Cra 突变导致肝脏中 IGF-1 的表达增加,全身 IGF-1 增加,最重要的是,脊髓 IGF-1 水平增加,这可能具有神经保护作用。
这些发现表明,动力蛋白基因中的 Cramping 突变对 SOD1(G93A)的保护至少部分是通过逆转能量不足和增加 IGF-1 向运动神经元的供应来介导的。