Pediatric Endocrinology and Diabetology, University Children's Hospital Bern, Bern, Switzerland.
PLoS One. 2011;6(5):e20178. doi: 10.1371/journal.pone.0020178. Epub 2011 May 27.
Steroidogenic acute regulatory protein (StAR) is crucial for transport of cholesterol to mitochondria where biosynthesis of steroids is initiated. Loss of StAR function causes lipoid congenital adrenal hyperplasia (LCAH).
StAR gene mutations causing partial loss of function manifest atypical and may be mistaken as familial glucocorticoid deficiency. Only a few mutations have been reported.
To report clinical, biochemical, genetic, protein structure and functional data on two novel StAR mutations, and to compare them with published literature.
Collaboration between the University Children's Hospital Bern, Switzerland, and the CIBERER, Hospital Vall d'Hebron, Autonomous University, Barcelona, Spain.
Two subjects of a non-consanguineous Caucasian family were studied. The 46,XX phenotypic normal female was diagnosed with adrenal insufficiency at the age of 10 months, had normal pubertal development and still has no signs of hypergonodatropic hypogonadism at 32 years of age. Her 46,XY brother was born with normal male external genitalia and was diagnosed with adrenal insufficiency at 14 months. Puberty was normal and no signs of hypergonadotropic hypogonadism are present at 29 years of age.
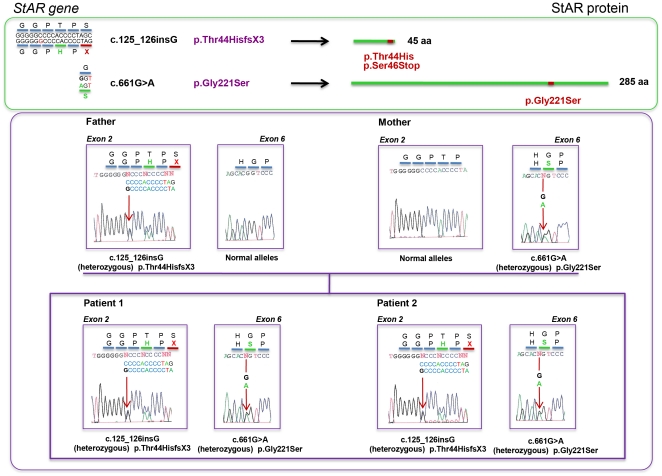
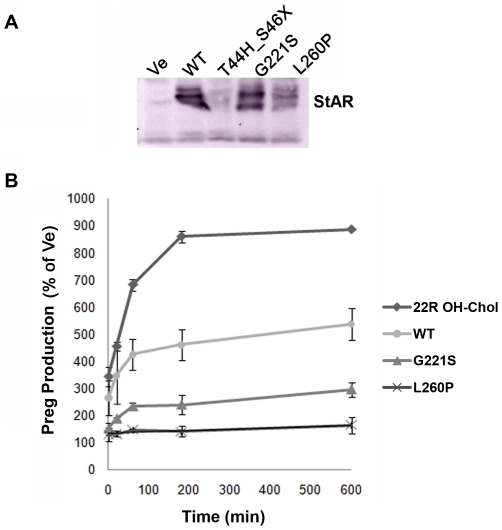
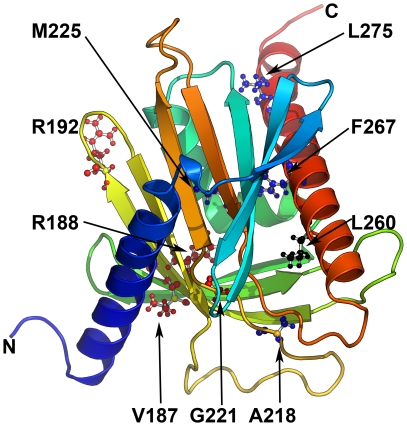
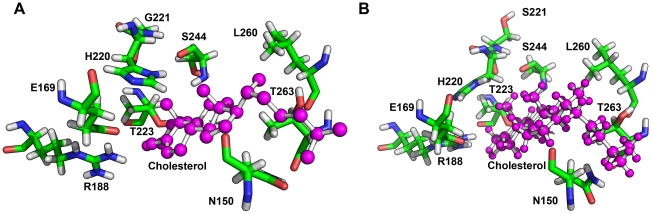
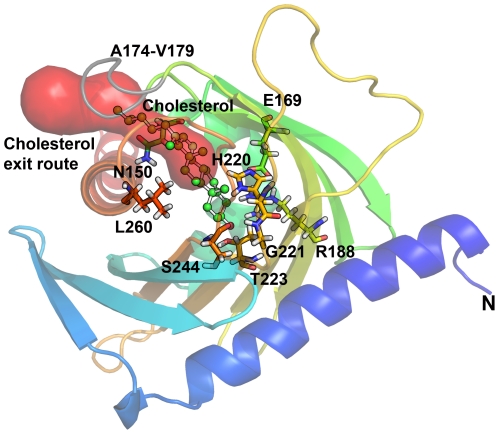
StAR gene analysis revealed two novel compound heterozygote mutations T44HfsX3 and G221S. T44HfsX3 is a loss-of-function StAR mutation. G221S retains partial activity (∼30%) and is therefore responsible for a milder, non-classic phenotype. G221S is located in the cholesterol binding pocket and seems to alter binding/release of cholesterol.
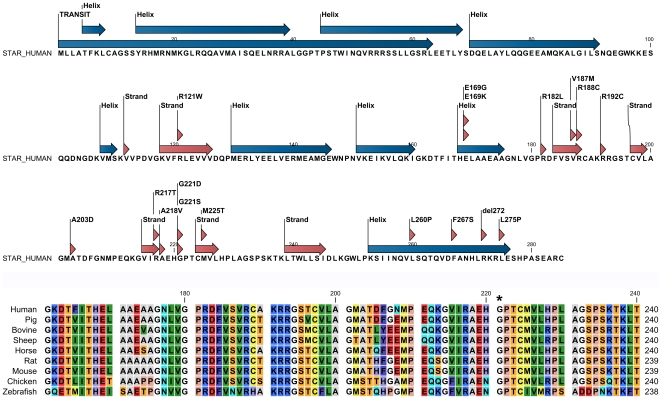
StAR mutations located in the cholesterol binding pocket (V187M, R188C, R192C, G221D/S) seem to cause non-classic lipoid CAH. Accuracy of genotype-phenotype prediction by in vitro testing may vary with the assays employed.
固醇急性调节蛋白(StAR)对于胆固醇向线粒体的转运至关重要,在那里启动类固醇的生物合成。StAR 功能丧失会导致脂质先天性肾上腺增生(LCAH)。
导致部分功能丧失的 StAR 基因突变表现为非典型,可能被误诊为家族性糖皮质激素缺乏症。仅有少数突变被报道。
报告两个新的 StAR 突变的临床、生化、遗传、蛋白质结构和功能数据,并与文献进行比较。
瑞士伯尔尼大学儿童医院和西班牙巴塞罗那自治大学 Vall d'Hebron 医院 CIBERER 之间的合作。
两个非近亲关系的白种人个体被研究。46,XX 表型正常的女性在 10 个月大时被诊断为肾上腺功能不全,青春期发育正常,32 岁时仍没有高促性腺激素性性腺功能减退的迹象。她的 46,XY 兄弟出生时外生殖器正常,在 14 个月时被诊断为肾上腺功能不全。青春期正常,29 岁时没有高促性腺激素性性腺功能减退的迹象。
StAR 基因分析显示两个新的复合杂合突变 T44HfsX3 和 G221S。T44HfsX3 是一种失活的 StAR 突变。G221S 保留部分活性(约 30%),因此导致较温和的非典型表型。G221S 位于胆固醇结合口袋中,似乎改变了胆固醇的结合/释放。
位于胆固醇结合口袋中的 StAR 突变(V187M、R188C、R192C、G221D/S)似乎导致非典型脂质先天性肾上腺增生。体外检测的基因型-表型预测的准确性可能因所使用的检测方法而异。