SSD Lab, Diagnosi Pre-Postnatale Malattie Metaboliche, IRCCS G, Gaslini, Genova, Italy.
Orphanet J Rare Dis. 2011 Jun 16;6:40. doi: 10.1186/1750-1172-6-40.
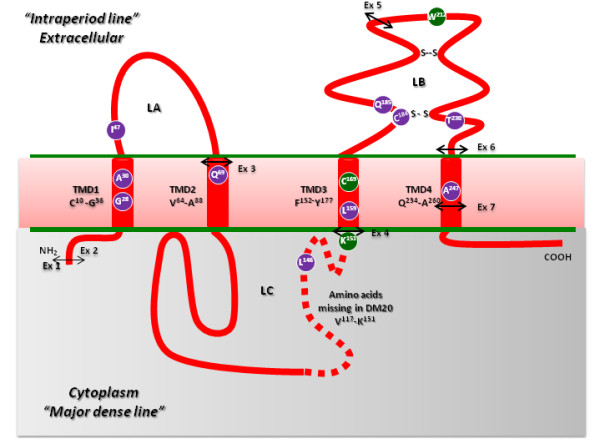
The breadth of the clinical spectrum underlying Pelizaeus-Merzbacher disease and spastic paraplegia type 2 is due to the extensive allelic heterogeneity in the X-linked PLP1 gene encoding myelin proteolipid protein (PLP). PLP1 mutations range from gene duplications of variable size found in 60-70% of patients to intragenic lesions present in 15-20% of patients.
Forty-eight male patients from 38 unrelated families with a PLP1-related disorder were studied. All DNA samples were screened for PLP1 gene duplications using real-time PCR. PLP1 gene sequencing analysis was performed on patients negative for the duplication. The mutational status of all 14 potential carrier mothers of the familial PLP1 gene mutation was determined as well as 15/24 potential carrier mothers of the PLP1 duplication.
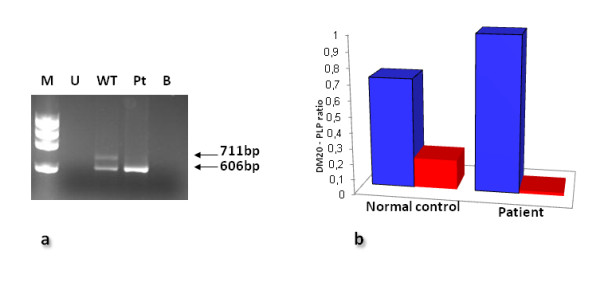
PLP1 gene duplications were identified in 24 of the unrelated patients whereas a variety of intragenic PLP1 mutations were found in the remaining 14 patients. Of the 14 different intragenic lesions, 11 were novel; these included one nonsense and 7 missense mutations, a 657-bp deletion, a microdeletion and a microduplication. The functional significance of the novel PLP1 missense mutations, all occurring at evolutionarily conserved residues, was analysed by the MutPred tool whereas their potential effect on splicing was ascertained using the Skippy algorithm and a neural network. Although MutPred predicted that all 7 novel missense mutations would be likely to be deleterious, in silico analysis indicated that four of them (p.Leu146Val, p.Leu159Pro, p.Thr230Ile, p.Ala247Asp) might cause exon skipping by altering exonic splicing elements. These predictions were then investigated in vitro for both p.Leu146Val and p.Thr230Ile by means of RNA or minigene studies and were subsequently confirmed in the case of p.Leu146Val. Peripheral neuropathy was noted in four patients harbouring intragenic mutations that altered RNA processing, but was absent from all PLP1-duplication patients. Unprecedentedly, family studies revealed the de novo occurrence of the PLP1 duplication at a frequency of 20%.
Pelizaeus-Merzbacher 病和痉挛性截瘫 2 型的临床表现广泛,这是由于 X 连锁 PLP1 基因编码髓鞘少突胶质细胞糖蛋白(PLP)的等位基因高度异质性所致。PLP1 突变范围从 60-70%患者中发现的大小可变的基因重复到 15-20%患者中存在的基因内病变。
研究了 38 个无关家庭的 48 名男性患者,这些患者均存在 PLP1 相关疾病。使用实时 PCR 对所有 DNA 样本进行 PLP1 基因重复筛查。对基因重复阴性的患者进行 PLP1 基因测序分析。还确定了所有 14 位有家族性 PLP1 基因突变的潜在女性携带者的突变状态,以及 24 位 PLP1 重复潜在女性携带者中的 15 位。
在 24 位无关患者中发现了 PLP1 基因重复,而在其余 14 位患者中发现了多种基因内 PLP1 突变。在 14 种不同的基因内病变中,有 11 种是新的;其中包括 1 种无义突变和 7 种错义突变、657bp 缺失、微缺失和微重复。使用 MutPred 工具分析了所有发生在进化上保守残基的新型 PLP1 错义突变的功能意义,使用 Skippy 算法和神经网络确定了它们对剪接的潜在影响。尽管 MutPred 预测所有 7 种新型错义突变都可能是有害的,但计算机分析表明,其中 4 种(p.Leu146Val、p.Leu159Pro、p.Thr230Ile、p.Ala247Asp)可能通过改变外显子剪接元件引起外显子跳跃。然后通过 RNA 或 minigene 研究对 p.Leu146Val 和 p.Thr230Ile 进行了体外研究,并在 p.Leu146Val 的情况下进行了证实。携带改变 RNA 处理的基因内突变的 4 名患者出现了周围神经病,但所有 PLP1 重复患者均无此症状。家族研究显示,PLP1 重复的发生率为 20%,这是前所未有的新生突变。