Queensland Facility for Advanced Bioinformatics, University of Queensland, 4072 St Lucia, QLD, Australia.
BMC Bioinformatics. 2011 Jun 22;12:253. doi: 10.1186/1471-2105-12-253.
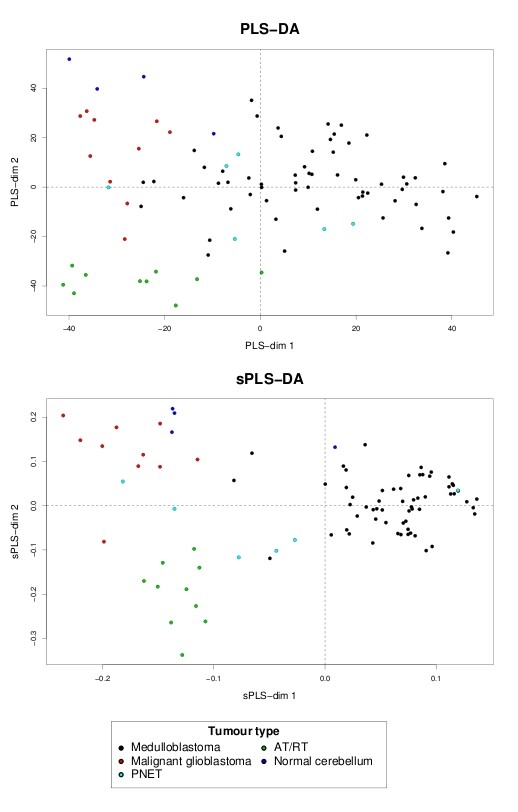
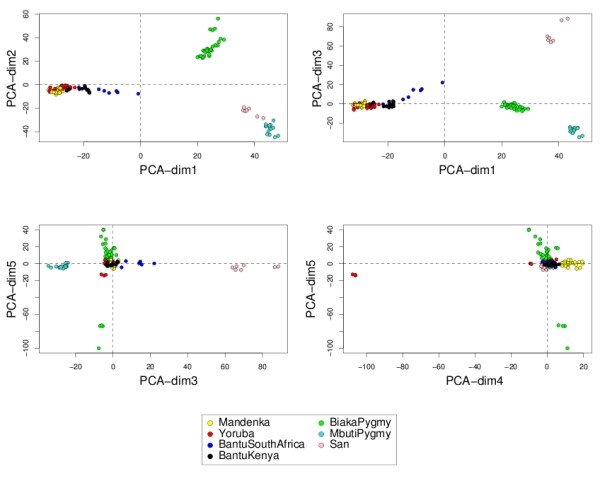
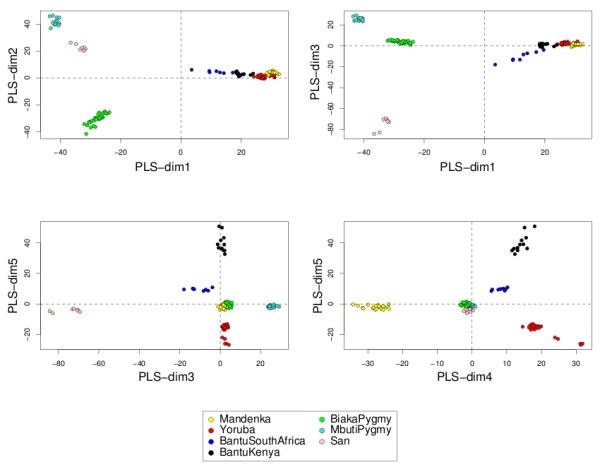
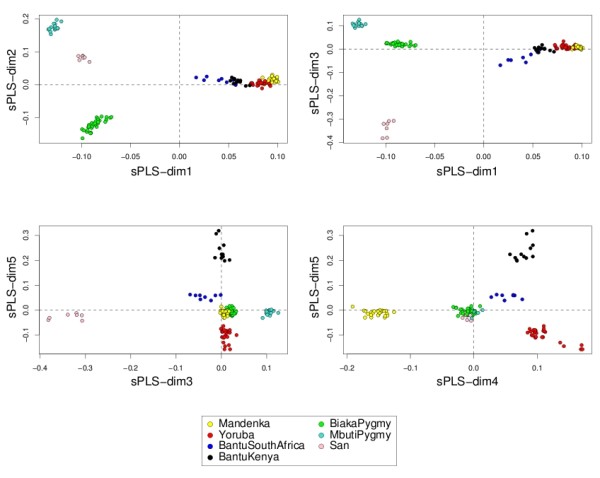
Variable selection on high throughput biological data, such as gene expression or single nucleotide polymorphisms (SNPs), becomes inevitable to select relevant information and, therefore, to better characterize diseases or assess genetic structure. There are different ways to perform variable selection in large data sets. Statistical tests are commonly used to identify differentially expressed features for explanatory purposes, whereas Machine Learning wrapper approaches can be used for predictive purposes. In the case of multiple highly correlated variables, another option is to use multivariate exploratory approaches to give more insight into cell biology, biological pathways or complex traits.
A simple extension of a sparse PLS exploratory approach is proposed to perform variable selection in a multiclass classification framework.
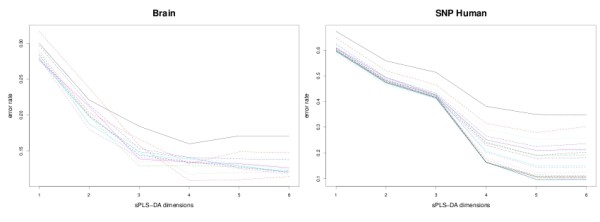
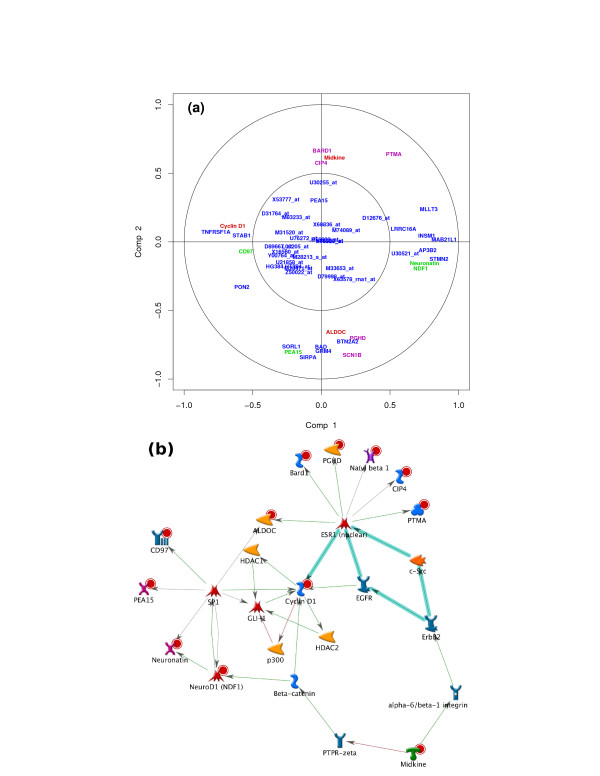
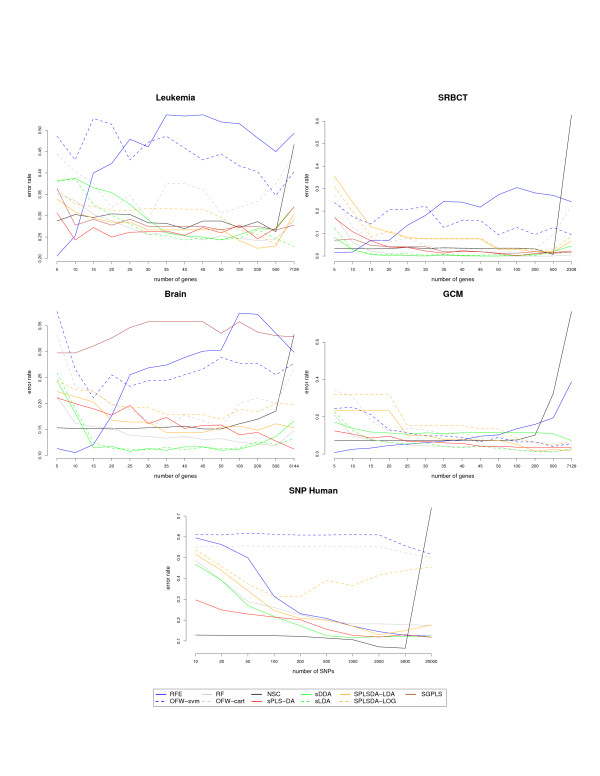
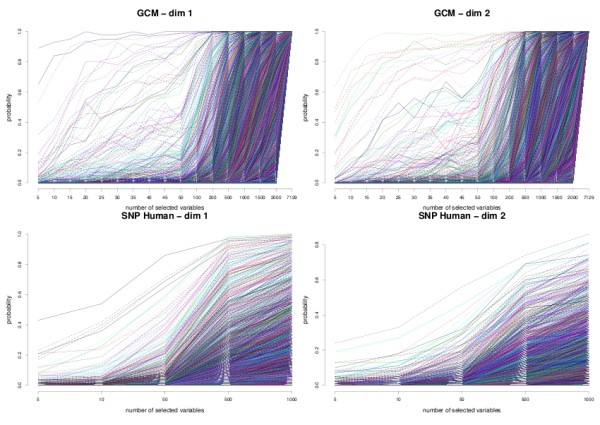
sPLS-DA has a classification performance similar to other wrapper or sparse discriminant analysis approaches on public microarray and SNP data sets. More importantly, sPLS-DA is clearly competitive in terms of computational efficiency and superior in terms of interpretability of the results via valuable graphical outputs. sPLS-DA is available in the R package mixOmics, which is dedicated to the analysis of large biological data sets.
在高通量生物数据(如基因表达或单核苷酸多态性 (SNP))上进行变量选择变得不可避免,以便选择相关信息,从而更好地描述疾病或评估遗传结构。在大型数据集上进行变量选择有不同的方法。统计检验常用于识别解释目的的差异表达特征,而机器学习包装器方法可用于预测目的。在多个高度相关变量的情况下,另一种选择是使用多元探索方法更深入地了解细胞生物学、生物途径或复杂特征。
提出了一种简单的稀疏 PLS 探索性方法的扩展,以在多类分类框架中进行变量选择。
sPLS-DA 在公共微阵列和 SNP 数据集上的分类性能与其他包装器或稀疏判别分析方法相似。更重要的是,sPLS-DA 在计算效率方面具有明显的竞争力,并且通过有价值的图形输出,在结果的可解释性方面具有优势。sPLS-DA 可在 R 包 mixOmics 中使用,该包专门用于分析大型生物数据集。