Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY, USA.
J Neuroinflammation. 2011 Jul 6;8:79. doi: 10.1186/1742-2094-8-79.
Overproduction of proinflammatory cytokines from activated microglia has been implicated as an important contributor to pathophysiology progression in both acute and chronic neurodegenerative diseases. Therefore, it is critical to elucidate intracellular signaling pathways that are significant contributors to cytokine overproduction in microglia exposed to specific stressors, especially pathways amenable to drug interventions. The serine/threonine protein kinase p38α MAPK is a key enzyme in the parallel and convergent intracellular signaling pathways involved in stressor-induced production of IL-1β and TNFα in peripheral tissues, and is a drug development target for peripheral inflammatory diseases. However, much less is known about the quantitative importance of microglial p38α MAPK in stressor-induced cytokine overproduction, or the potential of microglial p38α MAPK to be a druggable target for CNS disorders. Therefore, we examined the contribution of microglial p38αMAPK to cytokine up-regulation, with a focus on the potential to suppress the cytokine increase by inhibition of the kinase with pharmacological or genetic approaches.
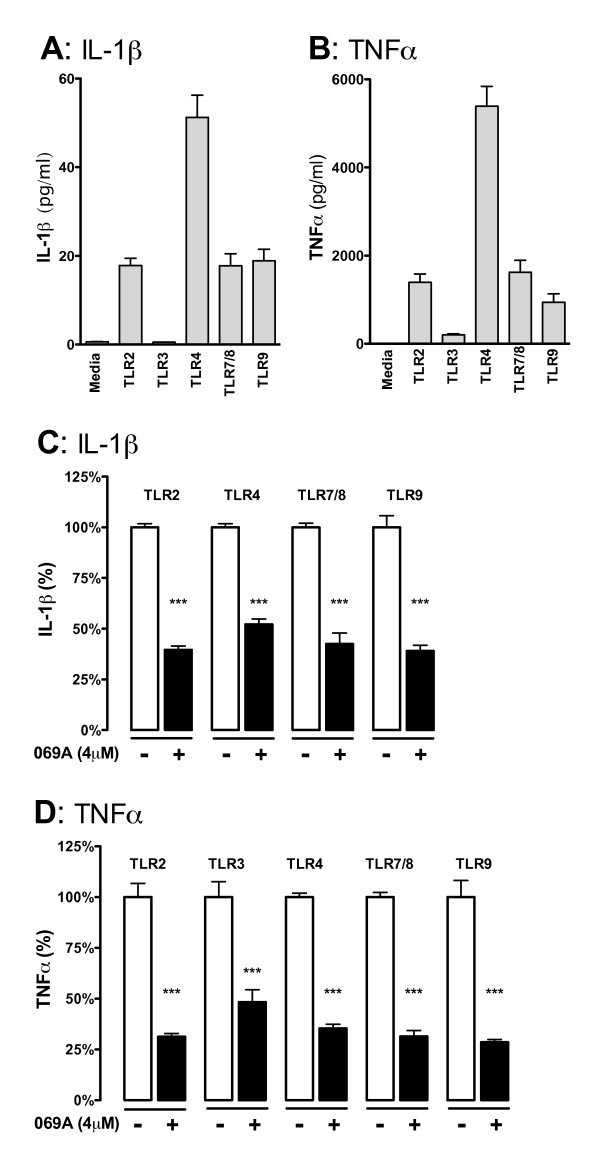
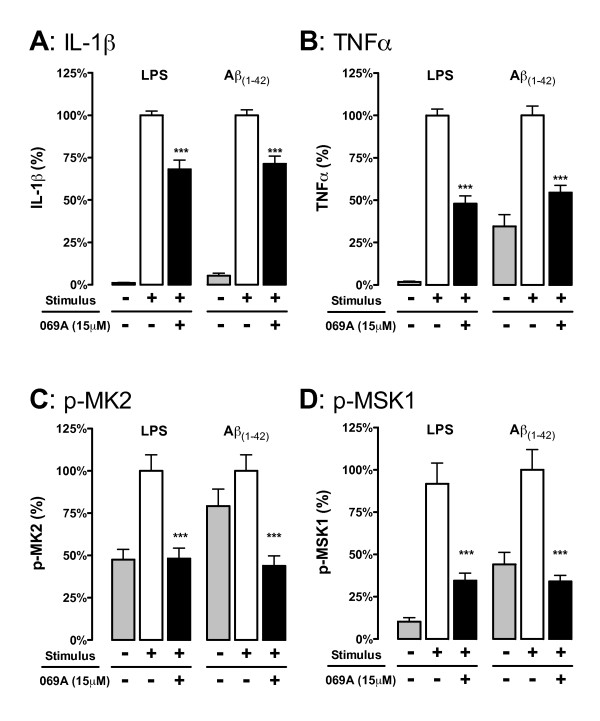
The microglial cytokine response to TLR ligands 2/3/4/7/8/9 or to Aβ1-42 was tested in the presence of a CNS-penetrant p38α MAPK inhibitor, MW01-2-069A-SRM. Primary microglia from mice genetically deficient in p38α MAPK were used to further establish a linkage between microglia p38α MAPK and cytokine overproduction. The in vivo significance was determined by p38α MAPK inhibitor treatment in a LPS-induced model of acute neuroinflammation.
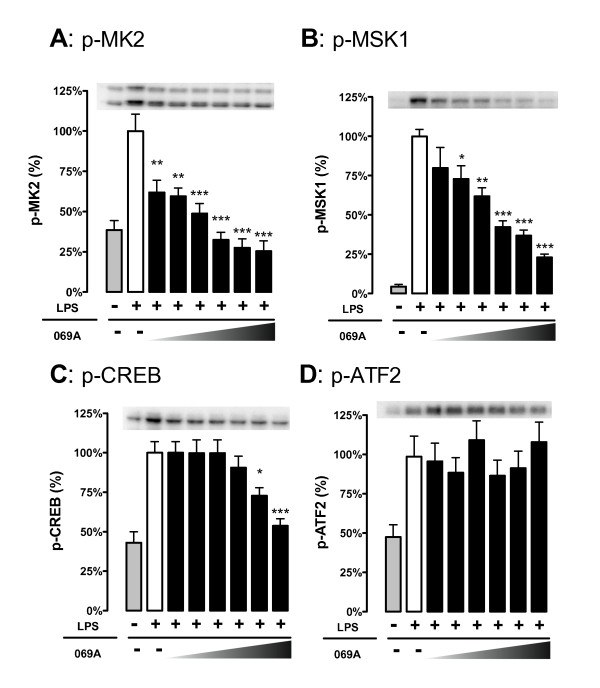
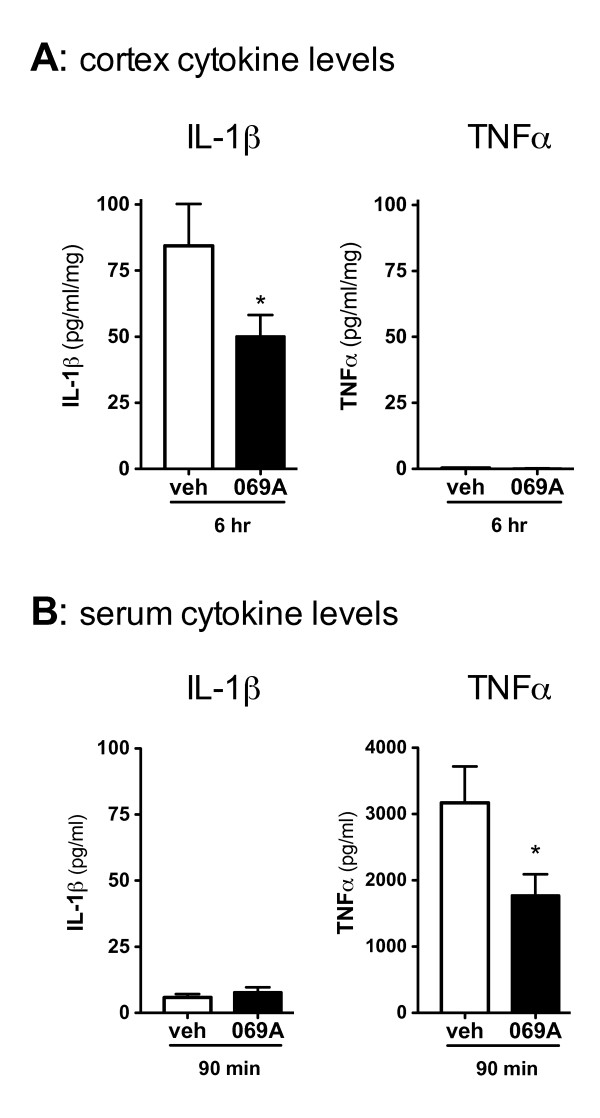
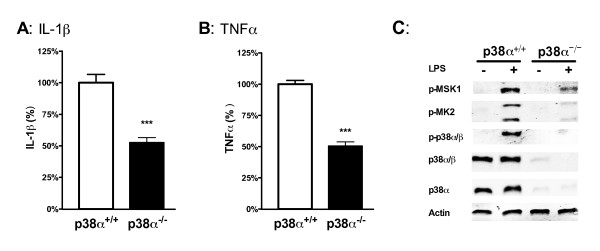
Increased IL-1β and TNFα production by the BV-2 microglial cell line and by primary microglia cultures was inhibited in a concentration-dependent manner by the p38α MAPK-targeted inhibitor. Cellular target engagement was demonstrated by the accompanying decrease in the phosphorylation state of two p38α MAPK protein substrates, MK2 and MSK1. Consistent with the pharmacological findings, microglia from p38α-deficient mice showed a diminished cytokine response to LPS. Further, oral administration of the inhibitor blocked the increase of IL-1β in the cerebral cortex of mice stressed by intraperitoneal injection of LPS.
The p38α MAPK pathway is an important contributor to the increased microglial production of proinflammatory cytokines induced by diverse stressors. The results also indicate the feasibility of targeting p38α MAPK to modulate CNS proinflammatory cytokine overproduction.
激活的小胶质细胞中促炎细胞因子的过度产生被认为是急性和慢性神经退行性疾病病理生理学进展的重要因素。因此,阐明小胶质细胞暴露于特定应激原时导致细胞因子过度产生的细胞内信号通路至关重要,尤其是那些可通过药物干预的信号通路。丝氨酸/苏氨酸蛋白激酶 p38α MAPK 是外周组织中应激诱导的 IL-1β 和 TNFα 产生的平行和会聚细胞内信号通路的关键酶,也是外周炎症性疾病的药物开发靶点。然而,对于小胶质细胞 p38α MAPK 在应激诱导的细胞因子过度产生中的定量重要性,或者小胶质细胞 p38α MAPK 是否有可能成为中枢神经系统疾病的可药物靶点,我们知之甚少。因此,我们研究了小胶质细胞 p38α MAPK 对细胞因子上调的贡献,重点关注通过药理学或遗传学方法抑制激酶来抑制细胞因子增加的潜力。
在中枢神经系统穿透性 p38α MAPK 抑制剂 MW01-2-069A-SRM 的存在下,检测 TLR 配体 2/3/4/7/8/9 或 Aβ1-42 对小胶质细胞的细胞因子反应。使用基因敲除 p38α MAPK 的小鼠的原代小胶质细胞进一步建立了小胶质细胞 p38α MAPK 与细胞因子过度产生之间的联系。通过 LPS 诱导的急性神经炎症模型中的 p38α MAPK 抑制剂治疗确定了体内的重要性。
BV-2 小胶质细胞系和原代小胶质细胞培养物中 IL-1β 和 TNFα 的产生增加,被 p38α MAPK 靶向抑制剂以浓度依赖性方式抑制。伴随的两个 p38α MAPK 蛋白底物 MK2 和 MSK1 的磷酸化状态的降低证明了细胞内靶标结合。与药理学发现一致,来自 p38α 缺陷型小鼠的小胶质细胞对 LPS 的细胞因子反应减弱。此外,口服给予抑制剂可阻断 LPS 腹腔注射应激小鼠大脑皮质中 IL-1β 的增加。
p38α MAPK 途径是多种应激原诱导的小胶质细胞产生促炎细胞因子增加的重要因素。结果还表明,靶向 p38α MAPK 以调节中枢神经系统促炎细胞因子过度产生是可行的。