Department of Medical Research, China Medical University Hospital, Taichung, Taiwan.
Genet Mol Biol. 2011 Apr;34(2):201-4. doi: 10.1590/s1415-47572011005000002. Epub 2011 Apr 1.
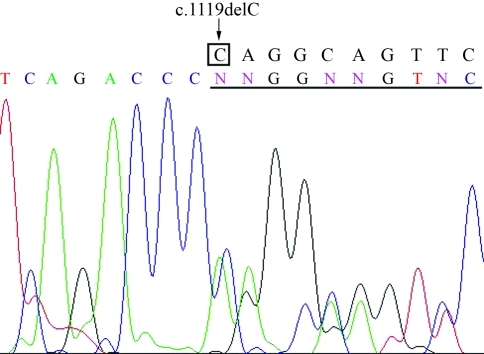
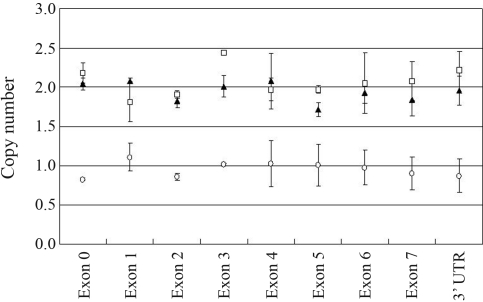
Cleidocranial dysplasia (CCD) is an autosomal dominant human skeletal disorder comprising hypoplastic clavicles, wide cranial sutures, supernumerary teeth, short stature, and other skeletal abnormalities. It is known that mutations in the human RUNX2 gene mapped at 6p21 are responsible for CCD. We analyzed the mutation patterns of the RUNX2 gene by direct sequencing in six Taiwanese index cases with typical CCD. One of the patients was a familial case and the others were sporadic cases. Sequencing identified four mutations. Three were caused by single nucleotide substitutions, which created a nonsense (p.R391X), two were missense mutations (p.R190W, p.R225Q), and the forth was a novel mutation (c.1119delC), a one-base deletion. Real time quantitative PCR adapted to determine copy numbers of the promoter, all exons and the 3'UTR region of the RUNX2 gene detected the deletion of a single allele in a sporadic case. The results extend the spectrum of RUNX2 mutations in CCD patients and indicate that complete deletions of the RUNX2 gene should be considered in those CCD patients lacking a point mutation detected by direct sequencing.
颅锁骨发育不全症(CCD)是一种常染色体显性遗传的人类骨骼疾病,其特征为锁骨发育不全、颅缝宽、额外牙齿、身材矮小和其他骨骼异常。已知位于 6p21 的人类 RUNX2 基因突变是 CCD 的致病原因。我们对六位有典型 CCD 的台湾籍先证者进行了 RUNX2 基因突变的直接测序分析。其中一位患者为家族性病例,其余为散发性病例。测序确定了四个突变。三个是由单核苷酸取代引起的,导致一个无义突变(p.R391X),两个是错义突变(p.R190W,p.R225Q),第四个是一个新的突变(c.1119delC),一个碱基缺失。实时定量 PCR 适用于检测 RUNX2 基因的启动子、所有外显子和 3'UTR 区域的拷贝数,在一个散发性病例中检测到一个等位基因的缺失。这些结果扩展了 CCD 患者 RUNX2 基因突变谱,并表明对于那些通过直接测序未检测到点突变的 CCD 患者,应考虑 RUNX2 基因的完全缺失。