Neuroadaptation Group, Max Planck Institute of Psychiatry, Munich, Germany.
PLoS One. 2011;6(7):e23097. doi: 10.1371/journal.pone.0023097. Epub 2011 Jul 29.
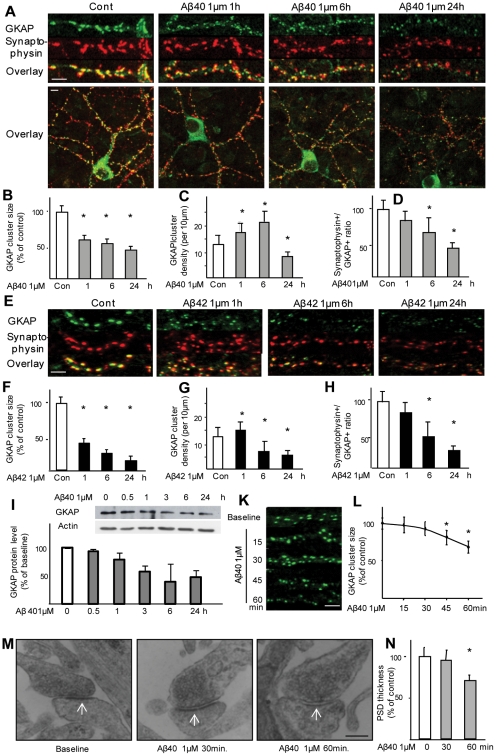
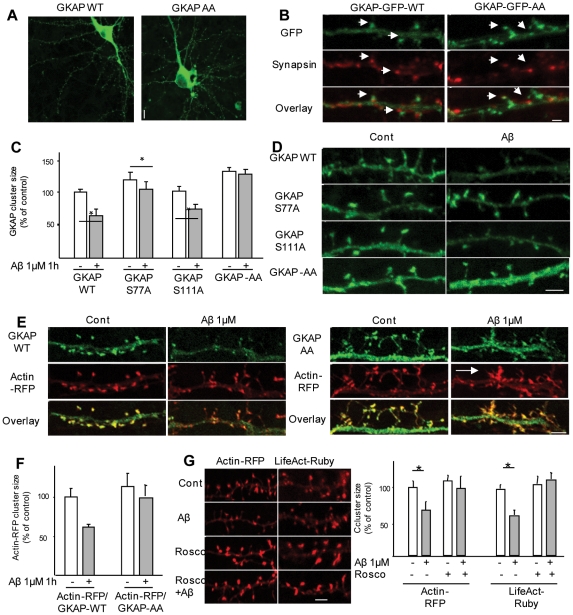
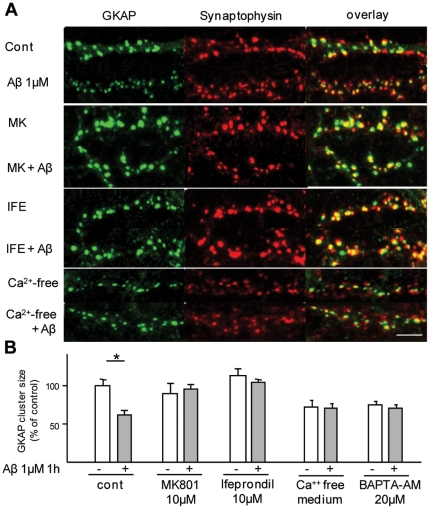
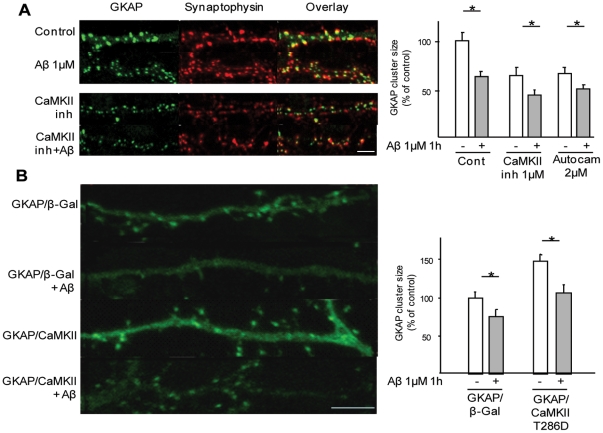
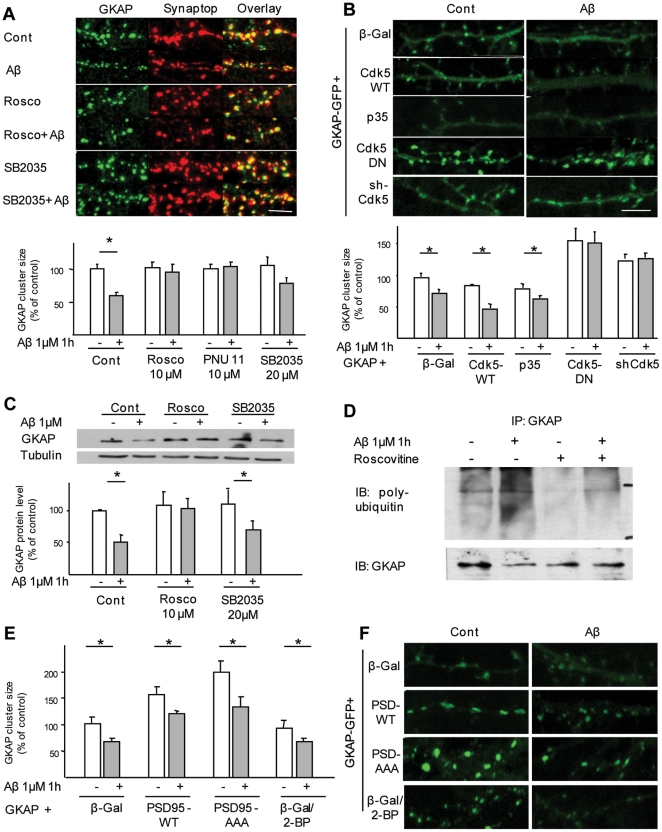
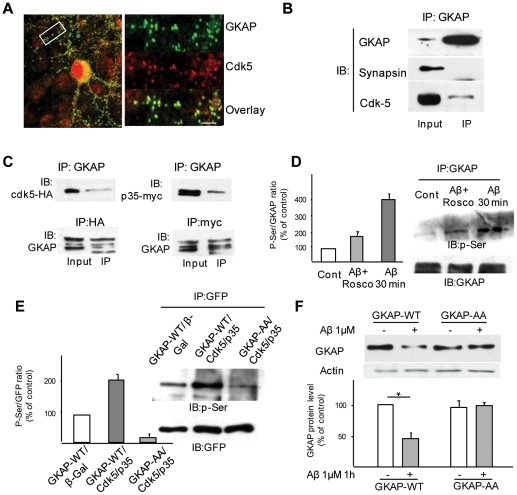
The early stages of Alzheimer's disease are marked by synaptic dysfunction and loss. This process results from the disassembly and degradation of synaptic components, in particular of scaffolding proteins that compose the post-synaptic density (PSD), namely PSD95, Homer and Shank. Here we investigated in rat frontal cortex dissociated culture the mechanisms involved in the downregulation of GKAP (SAPAP1), which links the PSD95 complex to the Shank complex and cytoskeletal structures within the PSD. We show that Aβ causes the rapid loss of GKAP from synapses through a pathway that critically requires cdk5 activity, and is set in motion by NMDAR activity and Ca(2+) influx. We show that GKAP is a direct substrate of cdk5 and that its phosphorylation results in polyubiquitination and proteasomal degradation of GKAP and remodeling (collapse) of the synaptic actin cytoskeleton; the latter effect is abolished in neurons expressing GKAP mutants that are resistant to phosphorylation by cdk5. Given that cdk5 also regulates degradation of PSD95, these results underscore the central position of cdk5 in mediating Aβ-induced PSD disassembly and synapse loss.
阿尔茨海默病的早期阶段以突触功能障碍和丧失为特征。这个过程源于突触成分的解体和降解,特别是组成突触后密度(PSD)的支架蛋白,即 PSD95、 Homer 和 Shank。在这里,我们在大鼠额皮质分离培养物中研究了与 GKAP(SAPAP1)下调相关的机制,GKAP 将 PSD95 复合物与 Shank 复合物和 PSD 内的细胞骨架结构联系起来。我们表明,Aβ 通过一条关键依赖于 cdk5 活性的途径导致 GKAP 从突触中迅速丢失,该途径由 NMDAR 活性和 Ca(2+)内流引发。我们表明 GKAP 是 cdk5 的直接底物,其磷酸化导致 GKAP 的多泛素化和蛋白酶体降解以及突触肌动蛋白细胞骨架的重塑(崩溃);在表达对 cdk5 磷酸化有抗性的 GKAP 突变体的神经元中,后一种效应被消除。鉴于 cdk5 还调节 PSD95 的降解,这些结果强调了 cdk5 在介导 Aβ 诱导的 PSD 解体和突触丢失中的核心地位。