Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, Indiana, United States of America.
PLoS One. 2011;6(8):e20988. doi: 10.1371/journal.pone.0020988. Epub 2011 Aug 2.
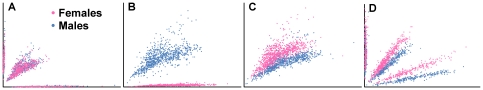
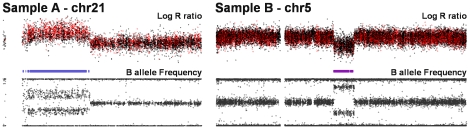
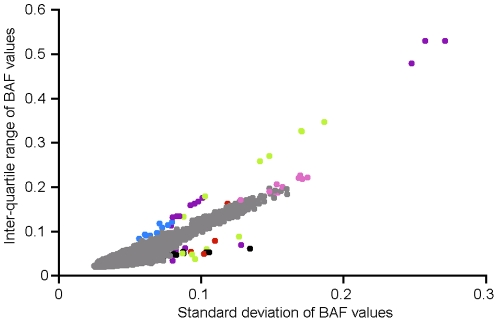
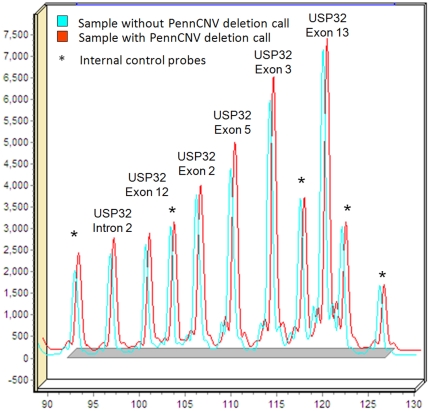
Copy number variants (CNVs) are known to cause Mendelian forms of Parkinson disease (PD), most notably in SNCA and PARK2. PARK2 has a recessive mode of inheritance; however, recent evidence demonstrates that a single CNV in PARK2 (but not a single missense mutation) may increase risk for PD. We recently performed a genome-wide association study for PD that excluded individuals known to have either a LRRK2 mutation or two PARK2 mutations. Data from the Illumina370Duo arrays were re-clustered using only white individuals with high quality intensity data, and CNV calls were made using two algorithms, PennCNV and QuantiSNP. After quality assessment, the final sample included 816 cases and 856 controls. Results varied between the two CNV calling algorithms for many regions, including the PARK2 locus (genome-wide p = 0.04 for PennCNV and p = 0.13 for QuantiSNP). However, there was consistent evidence with both algorithms for two novel genes, USP32 and DOCK5 (empirical, genome-wide p-values<0.001). PARK2 CNVs tended to be larger, and all instances that were molecularly tested were validated. In contrast, the CNVs in both novel loci were smaller and failed to replicate using real-time PCR, MLPA, and gel electrophoresis. The DOCK5 variation is more akin to a VNTR than a typical CNV and the association is likely caused by artifact due to DNA source. DNA for all the cases was derived from whole blood, while the DNA for all controls was derived from lymphoblast cell lines. The USP32 locus contains many SNPs with low minor allele frequency leading to a loss of heterozygosity that may have been spuriously interpreted by the CNV calling algorithms as support for a deletion. Thus, only the CNVs within the PARK2 locus could be molecularly validated and associated with PD susceptibility.
拷贝数变异(CNVs)已知可导致孟德尔形式的帕金森病(PD),尤其是在 SNCA 和 PARK2 中。 PARK2 具有隐性遗传模式;然而,最近的证据表明,PARK2 中的单个 CNV(而不是单个错义突变)可能会增加 PD 的风险。我们最近对 PD 进行了全基因组关联研究,排除了已知具有 LRRK2 突变或两个 PARK2 突变的个体。使用仅具有高质量强度数据的白人个体重新对 Illumina370Duo 阵列的数据进行聚类,并使用两种算法 PennCNV 和 QuantiSNP 进行 CNV 调用。经过质量评估,最终样本包括 816 例病例和 856 例对照。对于许多区域,包括 PARK2 基因座,两种 CNV 调用算法的结果均有所不同(PennCNV 的全基因组 p=0.04,QuantiSNP 的 p=0.13)。然而,两种算法都有一致的证据表明两个新基因 USP32 和 DOCK5 存在(经验,全基因组 p 值<0.001)。PARK2 CNVs 往往更大,并且所有经过分子检测的实例均得到验证。相比之下,两个新基因座中的 CNVs 较小,并且使用实时 PCR、MLPA 和凝胶电泳未能复制。DOCK5 变异更类似于 VNTR 而不是典型的 CNV,并且这种关联可能是由于 DNA 来源的artifact 引起的。所有病例的 DNA 均来自全血,而所有对照的 DNA 均来自淋巴母细胞系。USP32 基因座包含许多 SNP,其次要等位基因频率较低,导致杂合性丧失,这可能被 CNV 调用算法错误地解释为支持缺失。因此,只有 PARK2 基因座内的 CNVs 可以通过分子验证并与 PD 易感性相关。