Mishima Hiroyuki, Sasaki Kensaku, Tanaka Masahiro, Tatebe Osamu, Yoshiura Koh-Ichiro
Department of Human Genetics, Nagasaki University Graduate School of Biomedical Sciences, 1-12-4 Sakamoto, Nagasaki, Nagasaki, Japan.
BMC Res Notes. 2011 Sep 8;4:331. doi: 10.1186/1756-0500-4-331.
In bioinformatics projects, scientific workflow systems are widely used to manage computational procedures. Full-featured workflow systems have been proposed to fulfil the demand for workflow management. However, such systems tend to be over-weighted for actual bioinformatics practices. We realize that quick deployment of cutting-edge software implementing advanced algorithms and data formats, and continuous adaptation to changes in computational resources and the environment are often prioritized in scientific workflow management. These features have a greater affinity with the agile software development method through iterative development phases after trial and error.Here, we show the application of a scientific workflow system Pwrake to bioinformatics workflows. Pwrake is a parallel workflow extension of Ruby's standard build tool Rake, the flexibility of which has been demonstrated in the astronomy domain. Therefore, we hypothesize that Pwrake also has advantages in actual bioinformatics workflows.
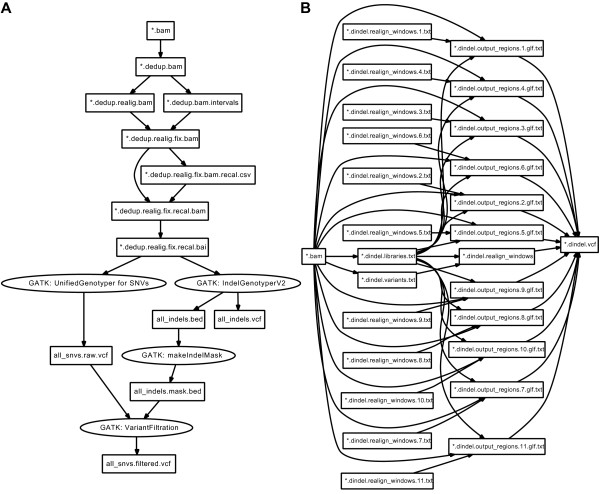
We implemented the Pwrake workflows to process next generation sequencing data using the Genomic Analysis Toolkit (GATK) and Dindel. GATK and Dindel workflows are typical examples of sequential and parallel workflows, respectively. We found that in practice, actual scientific workflow development iterates over two phases, the workflow definition phase and the parameter adjustment phase. We introduced separate workflow definitions to help focus on each of the two developmental phases, as well as helper methods to simplify the descriptions. This approach increased iterative development efficiency. Moreover, we implemented combined workflows to demonstrate modularity of the GATK and Dindel workflows.
Pwrake enables agile management of scientific workflows in the bioinformatics domain. The internal domain specific language design built on Ruby gives the flexibility of rakefiles for writing scientific workflows. Furthermore, readability and maintainability of rakefiles may facilitate sharing workflows among the scientific community. Workflows for GATK and Dindel are available at http://github.com/misshie/Workflows.
在生物信息学项目中,科学工作流系统被广泛用于管理计算过程。为满足工作流管理需求,人们提出了功能齐全的工作流系统。然而,这类系统对于实际的生物信息学实践而言往往过于臃肿。我们意识到,在科学工作流管理中,快速部署实现先进算法和数据格式的前沿软件,以及持续适应计算资源和环境的变化通常更为重要。通过反复试验后的迭代开发阶段,这些特性与敏捷软件开发方法具有更高的契合度。在此,我们展示了科学工作流系统Pwrake在生物信息学工作流中的应用。Pwrake是Ruby标准构建工具Rake的并行工作流扩展,其灵活性已在天文学领域得到证明。因此,我们假设Pwrake在实际的生物信息学工作流中也具有优势。
我们使用基因组分析工具包(GATK)和Dindel实现了Pwrake工作流来处理下一代测序数据。GATK和Dindel工作流分别是顺序工作流和并行工作流的典型示例。我们发现在实践中,实际的科学工作流开发在两个阶段进行迭代,即工作流定义阶段和参数调整阶段。我们引入了单独的工作流定义来帮助专注于这两个开发阶段中的每一个,以及辅助方法来简化描述。这种方法提高了迭代开发效率。此外,我们实现了组合工作流以展示GATK和Dindel工作流的模块化。
Pwrake能够对生物信息学领域的科学工作流进行敏捷管理。基于Ruby构建的内部领域特定语言设计赋予了rakefile编写科学工作流的灵活性。此外,rakefile的可读性和可维护性可能有助于在科学界共享工作流。GATK和Dindel的工作流可在http://github.com/misshie/Workflows获取。