Center for Computational Science, University of Miami, Miami, FL 33136, USA.
J Comput Aided Mol Des. 2011 Sep;25(9):873-83. doi: 10.1007/s10822-011-9469-2. Epub 2011 Sep 9.
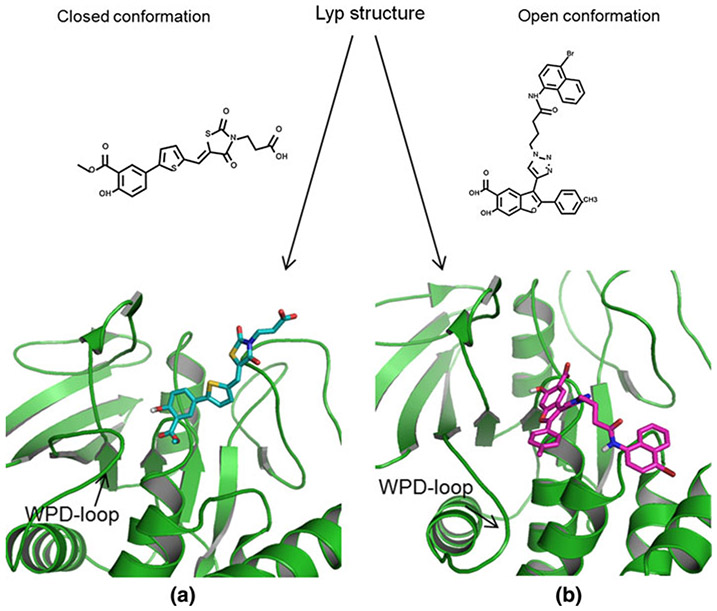
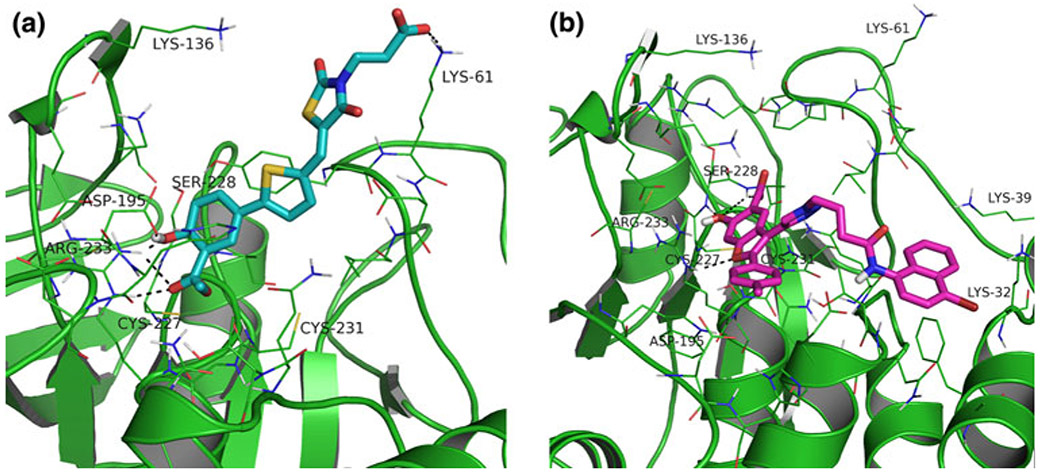
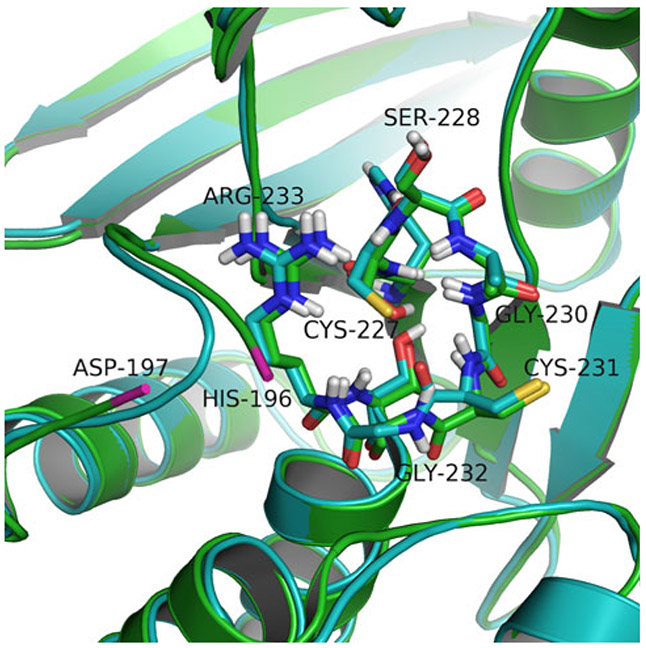
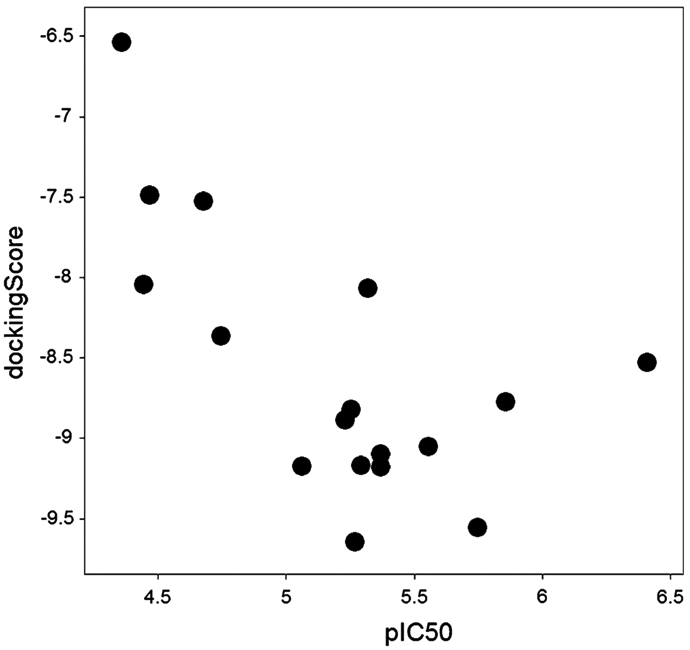
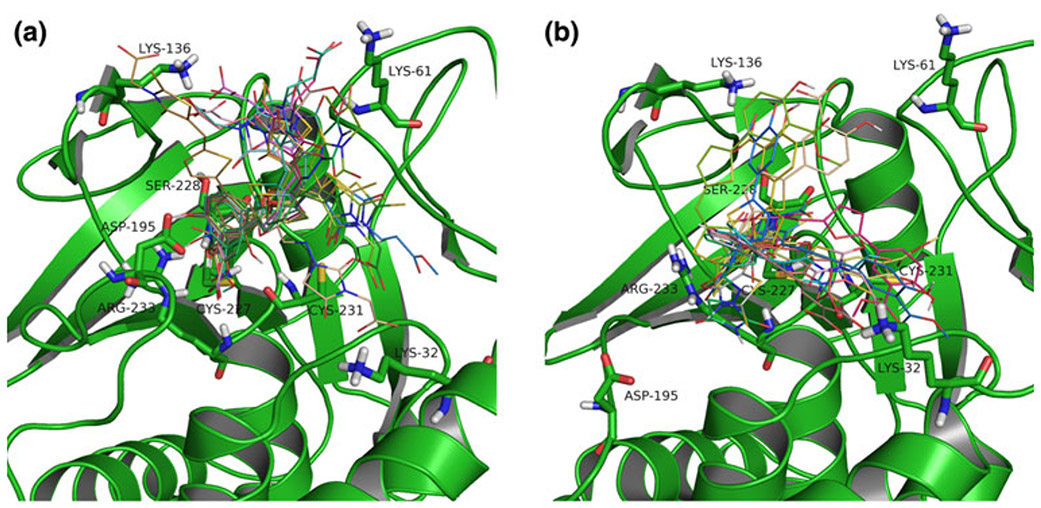
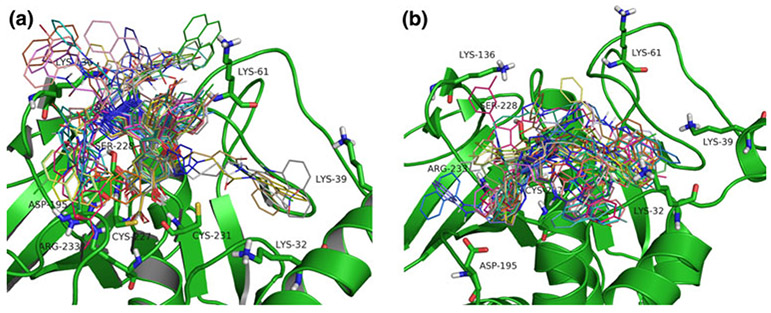
The lymphoid tyrosine phosphatase (LYP), encoded by the PTPN22 gene, has recently been identified as a promising drug target for human autoimmunity diseases. Like the majority of protein-tyrosine phosphatases LYP can adopt two functionally distinct forms determined by the conformation of the WPD-loop. The WPD-loop plays an important role in the catalytic dephosphorylation by protein-tyrosine phosphatases. Here we investigate the binding modes of two chemotypes of small molecule LYP inhibitors with respect to both protein conformations using computational modeling. To evaluate binding in the active form, we built a LYP protein structure model of high quality. Our results suggest that the two different compound classes investigated, bind to different conformations of the LYP phosphatase domain. Binding to the closed form is facilitated by an interaction with Asp195 in the WPD-loop, presumably stabilizing the active conformation. The analysis presented here is relevant for the design of inhibitors that specifically target either the closed or the open conformation of LYP in order to achieve better selectivity over phosphatases with similar binding sites.
淋巴特异酪氨酸磷酸酶(LYP)由 PTPN22 基因编码,最近被鉴定为人类自身免疫性疾病有前途的药物靶点。与大多数蛋白酪氨酸磷酸酶一样,LYP 可以采用两种功能上不同的形式,这取决于 WPD 环的构象。WPD 环在蛋白酪氨酸磷酸酶的催化去磷酸化中起着重要作用。在这里,我们使用计算建模研究了两种小分子 LYP 抑制剂的化学型在两种蛋白质构象下的结合模式。为了评估在活性形式下的结合,我们构建了一个高质量的 LYP 蛋白结构模型。我们的结果表明,所研究的两种不同的化合物类,结合到 LYP 磷酸酶结构域的不同构象。与 WPD 环中的 Asp195 相互作用促进了与封闭构象的结合,可能稳定了活性构象。这里提出的分析对于设计专门针对 LYP 的封闭或开放构象的抑制剂是相关的,以实现对具有相似结合位点的磷酸酶的更好选择性。