Department of Pharmacology, Institute of Clinical Medicine, Faculty of Medicine and Oslo University Hospital, University of Oslo, Oslo, Norway.
BMC Cancer. 2011 Oct 2;11:421. doi: 10.1186/1471-2407-11-421.
Neurotensin has been found to promote colon carcinogenesis in rats and mice, and proliferation of human colon carcinoma cell lines, but the mechanisms involved are not clear. We have examined signalling pathways activated by neurotensin in colorectal and pancreatic carcinoma cells.
Colon carcinoma cell lines HCT116 and HT29 and pancreatic adenocarcinoma cell line Panc-1 were cultured and stimulated with neurotensin or epidermal growth factor (EGF). DNA synthesis was determined by incorporation of radiolabelled thymidine into DNA. Levels and phosphorylation of proteins in signalling pathways were assessed by Western blotting.
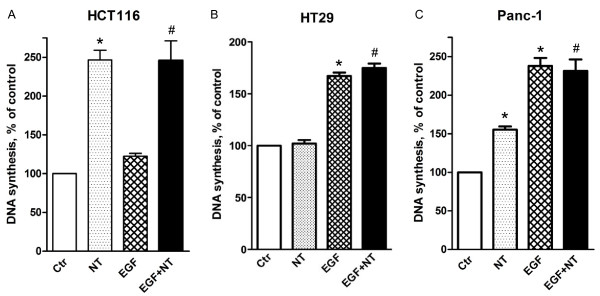
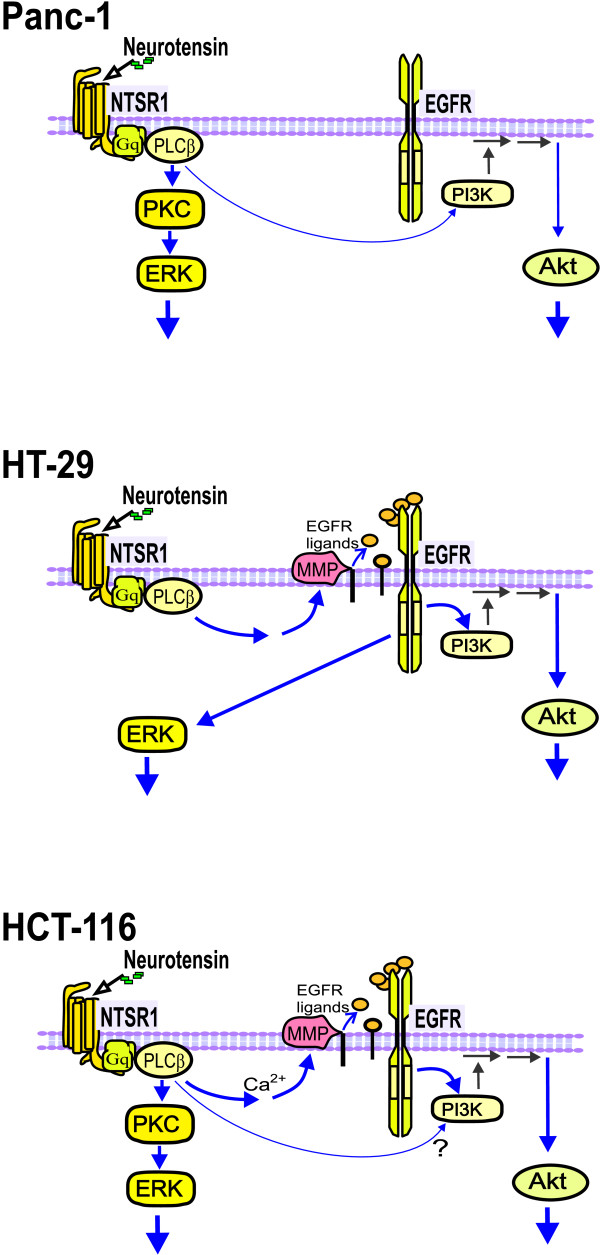
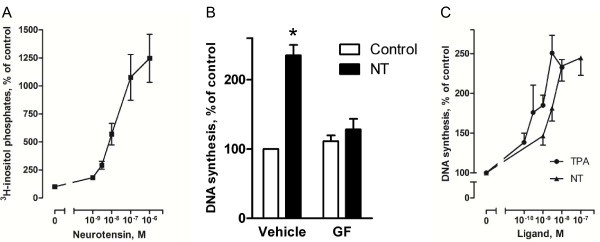
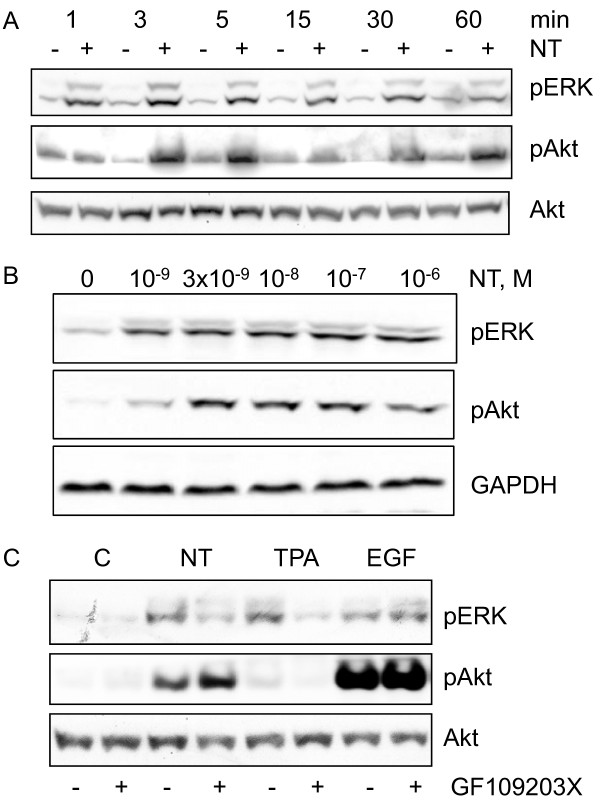
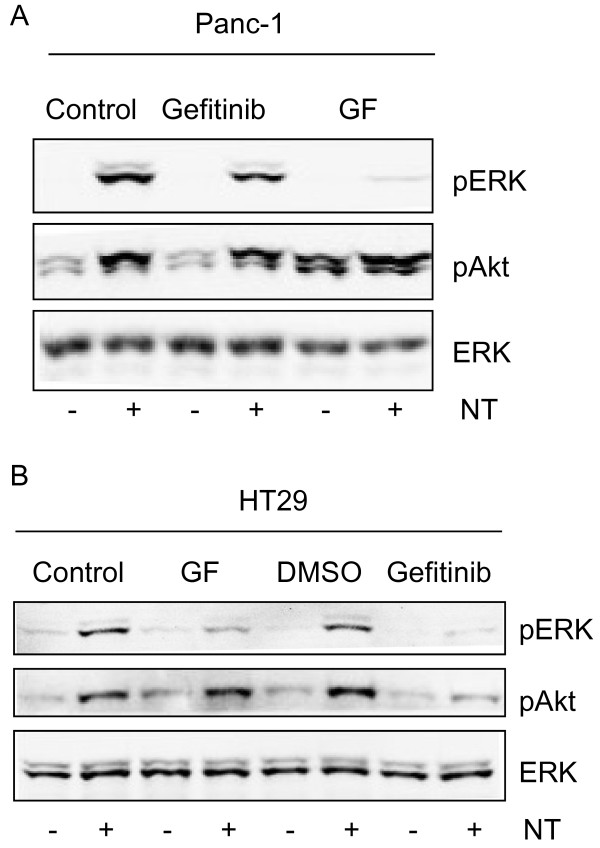
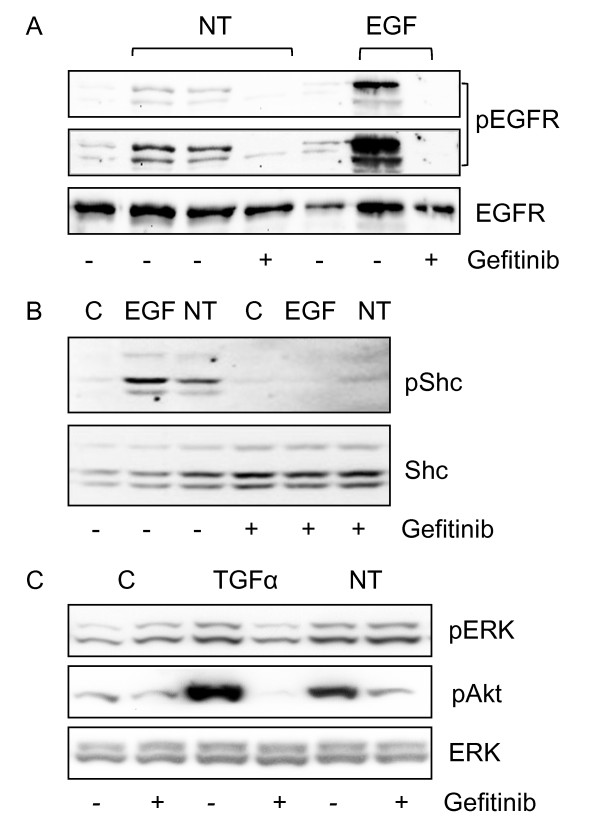
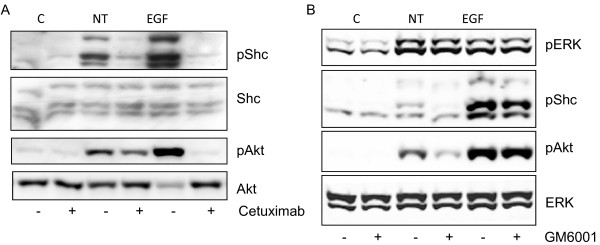
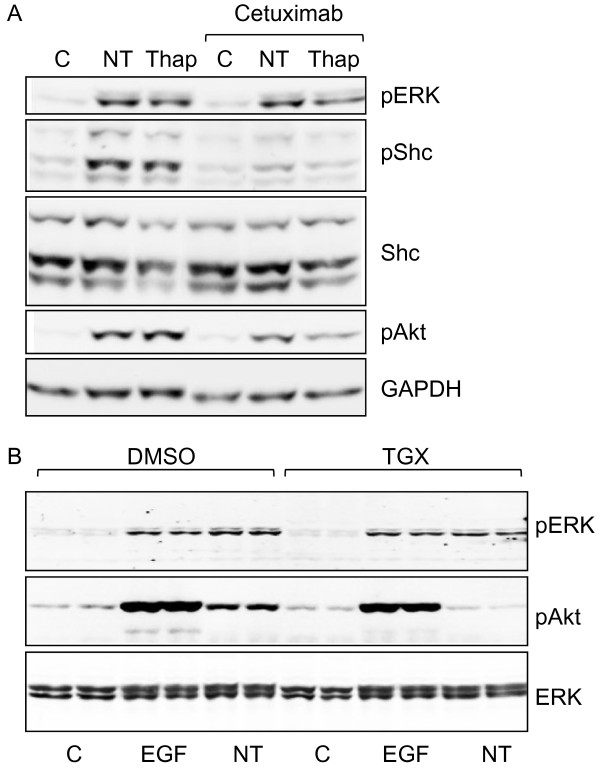
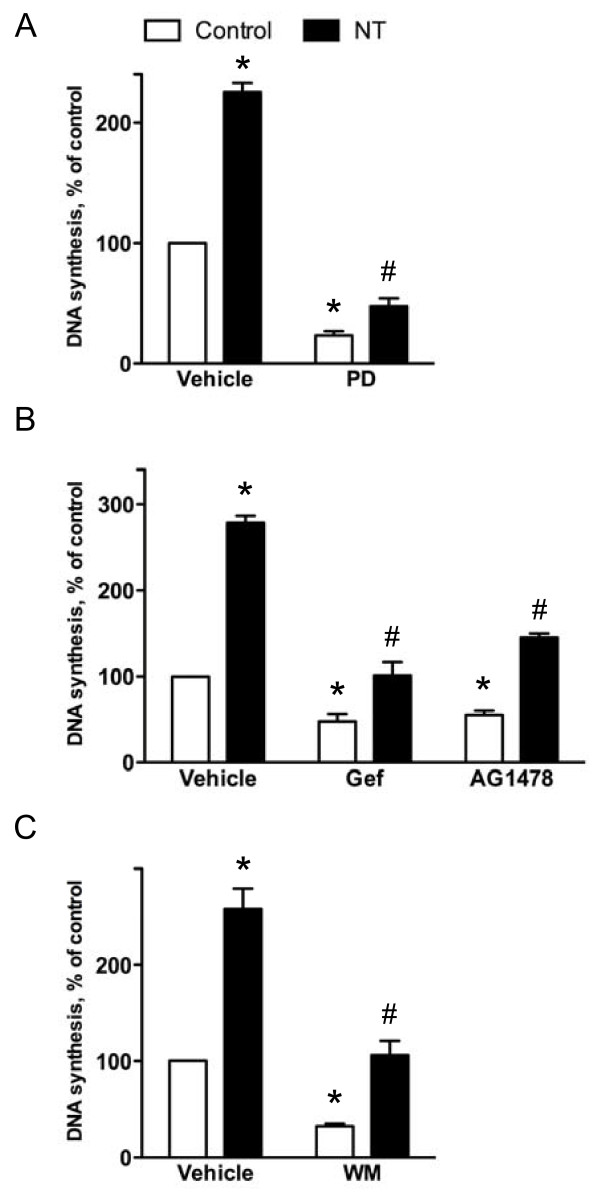
Neurotensin stimulated the phosphorylation of both extracellular signal-regulated kinase (ERK) and Akt in all three cell lines, but apparently did so through different pathways. In Panc-1 cells, neurotensin-induced phosphorylation of ERK, but not Akt, was dependent on protein kinase C (PKC), whereas an inhibitor of the β-isoform of phosphoinositide 3-kinase (PI3K), TGX221, abolished neurotensin-induced Akt phosphorylation in these cells, and there was no evidence of EGF receptor (EGFR) transactivation. In HT29 cells, in contrast, the EGFR tyrosine kinase inhibitor gefitinib blocked neurotensin-stimulated phosphorylation of both ERK and Akt, indicating transactivation of EGFR, independently of PKC. In HCT116 cells, neurotensin induced both a PKC-dependent phosphorylation of ERK and a metalloproteinase-mediated transactivation of EGFR that was associated with a gefitinib-sensitive phosphorylation of the downstream adaptor protein Shc. The activation of Akt was also inhibited by gefitinib, but only partly, suggesting a mechanism in addition to EGFR transactivation. Inhibition of PKC blocked neurotensin-induced DNA synthesis in HCT116 cells.
While acting predominantly through PKC in Panc-1 cells and via EGFR transactivation in HT29 cells, neurotensin used both these pathways in HCT116 cells. In these cells, neurotensin-induced activation of ERK and stimulation of DNA synthesis was PKC-dependent, whereas activation of the PI3K/Akt pathway was mediated by stimulation of metalloproteinases and subsequent transactivation of the EGFR. Thus, the data show that the signalling mechanisms mediating the effects of neurotensin involve multiple pathways and are cell-dependent.
神经降压素已被发现可促进大鼠和小鼠的结肠癌发生,并促进人结肠癌细胞系的增殖,但涉及的机制尚不清楚。我们研究了神经降压素在结直肠和胰腺癌细胞中的信号通路。
培养结肠癌细胞系 HCT116 和 HT29 以及胰腺腺癌细胞系 Panc-1,并使用神经降压素或表皮生长因子(EGF)刺激它们。通过放射性标记的胸苷掺入 DNA 来测定 DNA 合成。通过 Western 印迹评估信号通路中蛋白质的水平和磷酸化。
神经降压素刺激所有三种细胞系中的细胞外信号调节激酶(ERK)和 Akt 的磷酸化,但显然是通过不同的途径。在 Panc-1 细胞中,神经降压素诱导的 ERK 磷酸化,但不是 Akt,依赖于蛋白激酶 C(PKC),而β-肌醇磷脂 3-激酶(PI3K)的抑制剂 TGX221 可消除这些细胞中神经降压素诱导的 Akt 磷酸化,并且没有证据表明表皮生长因子受体(EGFR)的转激活。相比之下,在 HT29 细胞中,EGFR 酪氨酸激酶抑制剂吉非替尼阻断了神经降压素刺激的 ERK 和 Akt 的磷酸化,表明 EGFR 的转激活,独立于 PKC。在 HCT116 细胞中,神经降压素诱导了 PKC 依赖性 ERK 磷酸化和金属蛋白酶介导的 EGFR 的转激活,这与 gefitinib 敏感的下游衔接蛋白 Shc 的磷酸化有关。Akt 的激活也被 gefitinib 抑制,但只是部分抑制,表明除了 EGFR 转激活之外还有其他机制。PKC 抑制阻断了 HCT116 细胞中神经降压素诱导的 DNA 合成。
虽然在 Panc-1 细胞中主要通过 PKC 起作用,在 HT29 细胞中通过 EGFR 转激活,但在 HCT116 细胞中神经降压素同时使用了这两种途径。在这些细胞中,神经降压素诱导的 ERK 激活和 DNA 合成刺激依赖于 PKC,而 PI3K/Akt 通路的激活则由金属蛋白酶的刺激和随后的 EGFR 转激活介导。因此,数据表明介导神经降压素作用的信号机制涉及多种途径并且依赖于细胞。