Larsen Jan, Pettersson Olof J, Jakobsen Maria, Thomsen Rune, Pedersen Christina B, Hertz Jens M, Gregersen Niels, Corydon Thomas J, Jensen Thomas G
Department of Biomedicine, Aarhus University, Aarhus, Denmark.
BMC Res Notes. 2011 Nov 11;4:490. doi: 10.1186/1756-0500-4-490.
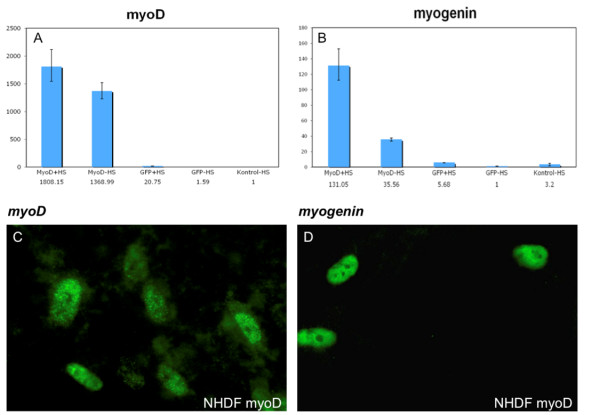
Myotonic dystrophy type 1 (DM1) is the most common muscle dystrophy in adults. The disease is caused by a triplet expansion in the 3'end of the myotonic dystrophy protein kinase (DMPK) gene. In order to develop a human cell model for investigation of possible effects of antisense and RNAi effector molecules we have used lentiviral mediated myoD-forced myogenesis of DM1 patient fibroblasts.
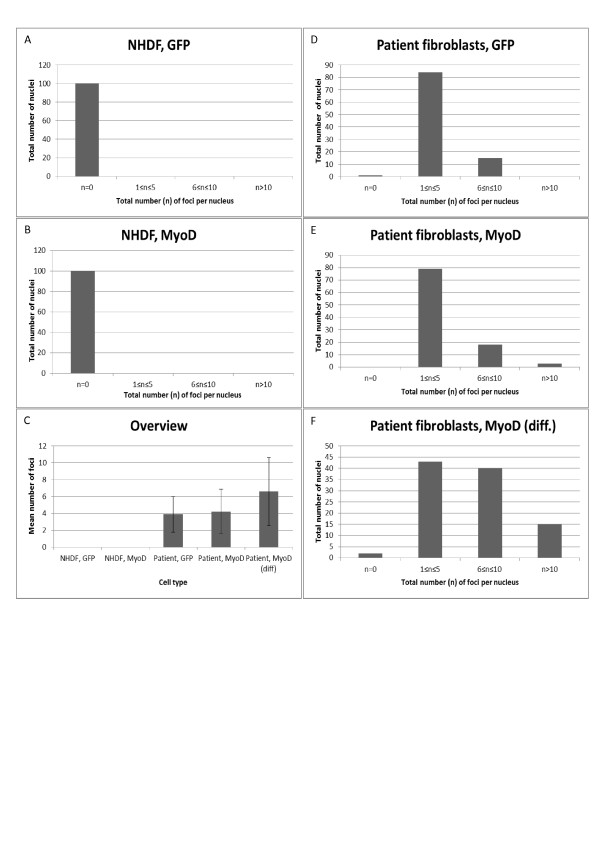
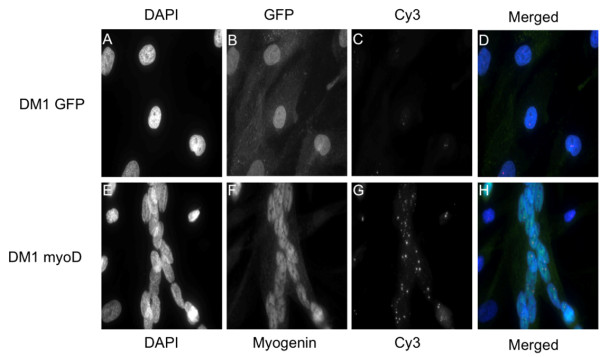
Transduced fibroblasts show a multinuclear phenotype and express the differentiation marker myogenin. Furthermore, fluorescence in situ hybridization (FISH) analysis revealed a statistical significant increase in the amount of nuclear foci in DM1 patient fibroblasts after myogenesis. Finally, no nuclear foci were found after treatment with oligonucleotides targeting the repeat expansions.
The abundance of nuclear foci in DM1 patient fibroblasts increase following myogenesis, as visualized by FISH analysis. Foci were eradicated after treatment with antisense oligonucleotides. Thus, we propose that the current cell model is suitable for testing of novel treatment modalities.
1型强直性肌营养不良(DM1)是成人中最常见的肌肉营养不良症。该疾病由强直性肌营养不良蛋白激酶(DMPK)基因3'端的三联体扩增引起。为了建立一个人类细胞模型来研究反义寡核苷酸和RNA干扰效应分子的潜在作用,我们利用慢病毒介导的DM1患者成纤维细胞的肌分化因子(myoD)诱导成肌作用。
转导后的成纤维细胞呈现多核表型并表达分化标志物肌细胞生成素。此外,荧光原位杂交(FISH)分析显示,成肌作用后DM1患者成纤维细胞核灶数量有统计学意义的增加。最后,用靶向重复扩增序列的寡核苷酸处理后未发现核灶。
FISH分析显示,DM1患者成纤维细胞成肌作用后核灶数量增加。反义寡核苷酸处理后核灶消失。因此,我们认为当前的细胞模型适用于测试新的治疗方法。