Center for Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI, USA.
Genet Epidemiol. 2011 Dec;35(8):766-80. doi: 10.1002/gepi.20626.
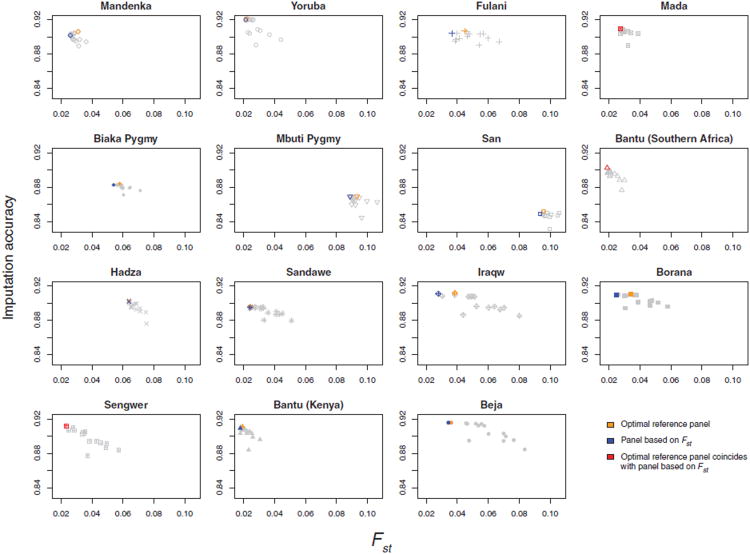
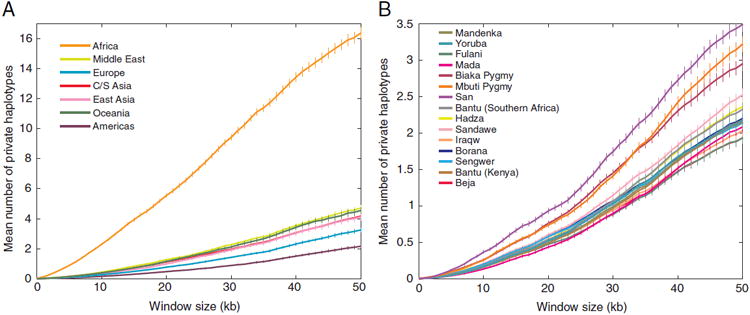
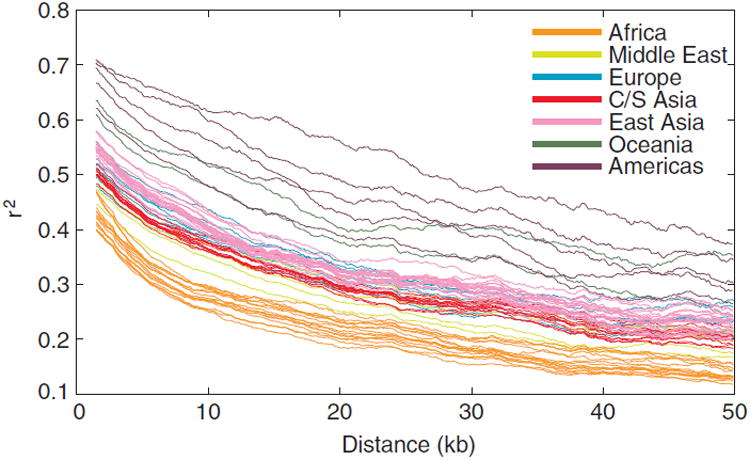
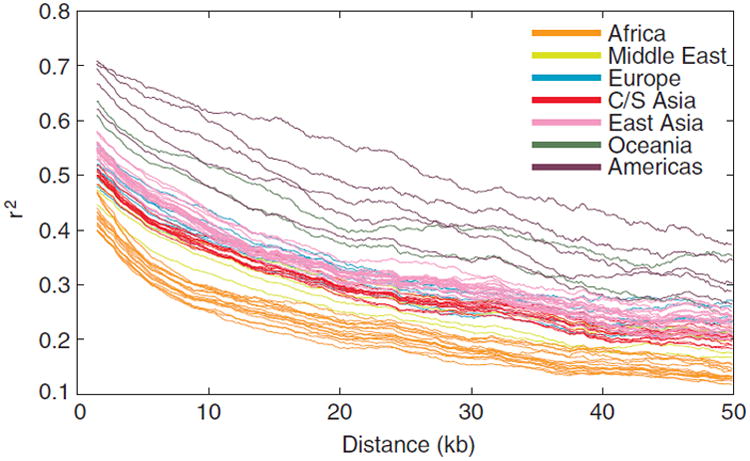
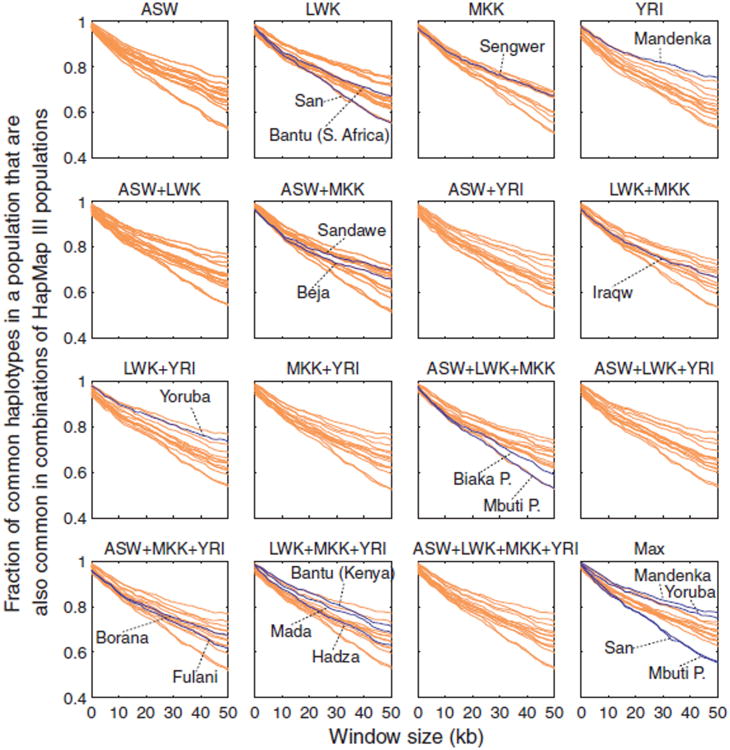
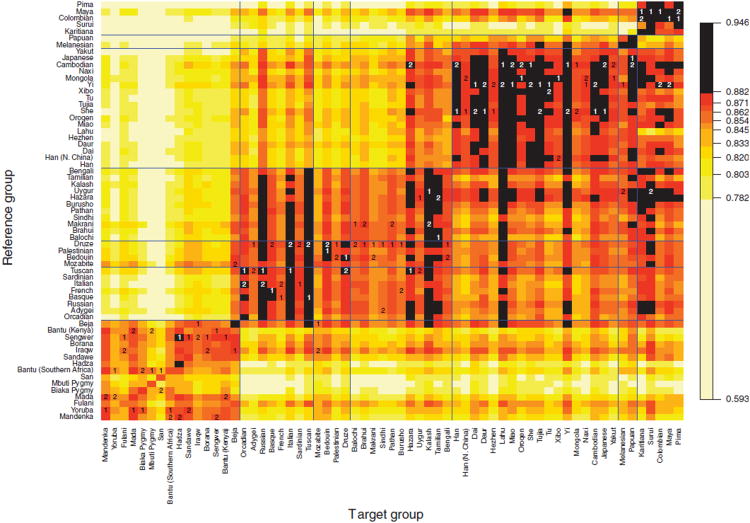
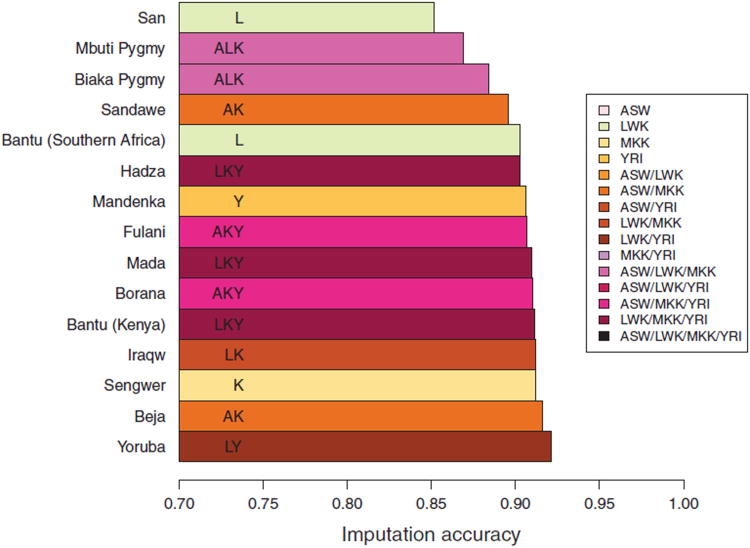
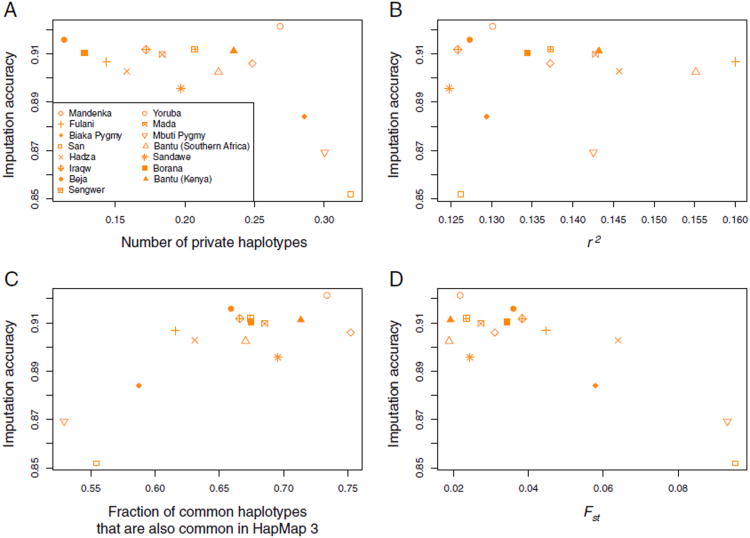
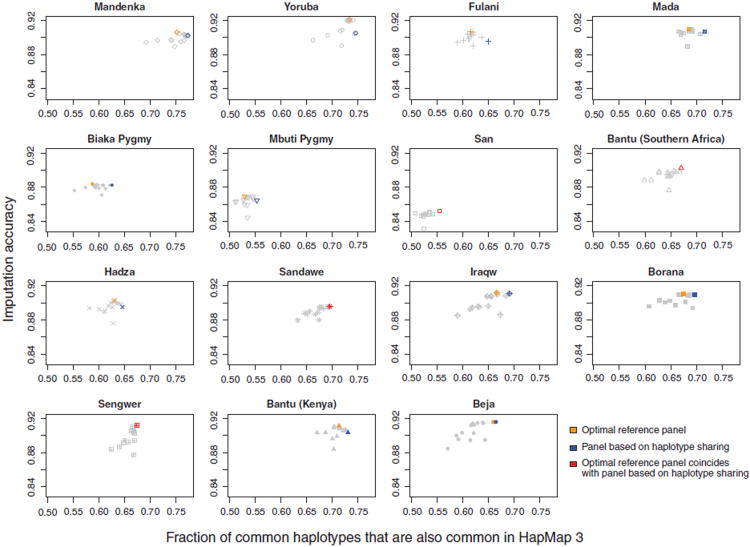
Sub-Saharan Africa has been identified as the part of the world with the greatest human genetic diversity. This high level of diversity causes difficulties for genome-wide association (GWA) studies in African populations-for example, by reducing the accuracy of genotype imputation in African populations compared to non-African populations. Here, we investigate haplotype variation and imputation in Africa, using 253 unrelated individuals from 15 Sub-Saharan African populations. We identify the populations that provide the greatest potential for serving as reference panels for imputing genotypes in the remaining groups. Considering reference panels comprising samples of recent African descent in Phase 3 of the HapMap Project, we identify mixtures of reference groups that produce the maximal imputation accuracy in each of the sampled populations. We find that optimal HapMap mixtures and maximal imputation accuracies identified in detailed tests of imputation procedures can instead be predicted by using simple summary statistics that measure relationships between the pattern of genetic variation in a target population and the patterns in potential reference panels. Our results provide an empirical basis for facilitating the selection of reference panels in GWA studies of diverse human populations, especially those of African ancestry.
撒哈拉以南非洲已被确定为世界上人类遗传多样性最大的地区。这种高度的多样性给非洲人群的全基因组关联(GWA)研究带来了困难——例如,与非非洲人群相比,它降低了非洲人群中基因型推断的准确性。在这里,我们使用来自 15 个撒哈拉以南非洲人群的 253 个无关个体来研究非洲的单倍型变异和推断。我们确定了最有潜力作为其余群体基因型推断参考面板的人群。考虑到 HapMap 项目第三阶段中具有近期非洲血统的样本组成的参考面板,我们确定了在每个抽样人群中产生最大推断准确性的参考组混合物。我们发现,在对推断程序进行详细测试中确定的最佳 HapMap 混合物和最大推断准确性,可以通过使用简单的汇总统计量来预测,这些统计量衡量目标人群中遗传变异模式与潜在参考面板模式之间的关系。我们的研究结果为促进不同人群,特别是具有非洲血统的人群的 GWA 研究中参考面板的选择提供了经验基础。