Department of Biochemistry and Molecular Genetics, School of Medicine, University of Colorado, Aurora, Colorado, USA.
PLoS Genet. 2011 Dec;7(12):e1002384. doi: 10.1371/journal.pgen.1002384. Epub 2011 Dec 1.
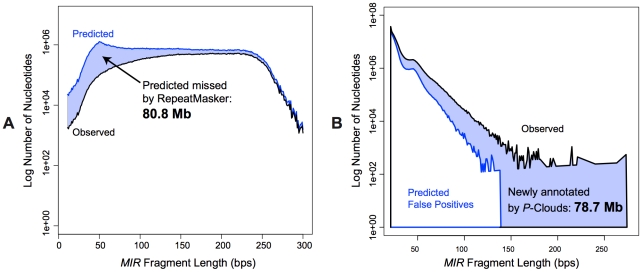
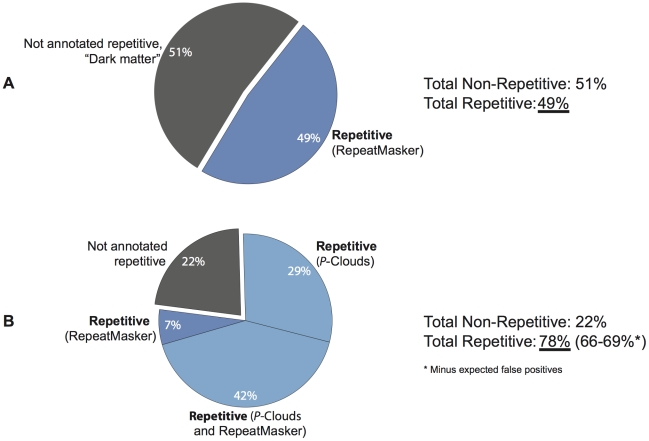
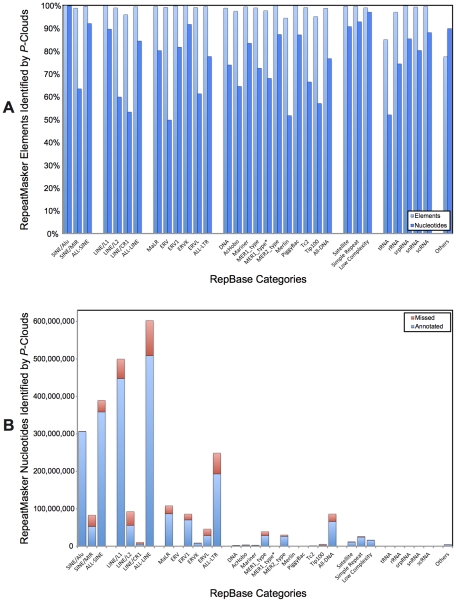
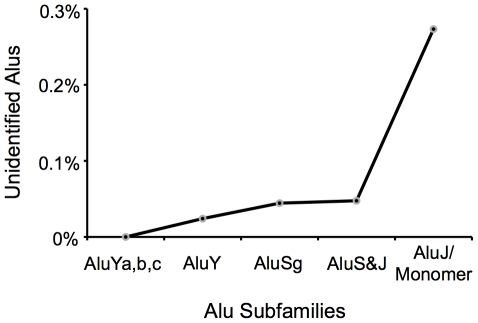
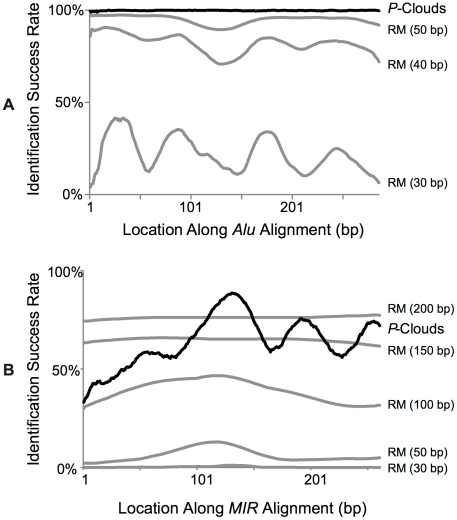
Transposable elements (TEs) are conventionally identified in eukaryotic genomes by alignment to consensus element sequences. Using this approach, about half of the human genome has been previously identified as TEs and low-complexity repeats. We recently developed a highly sensitive alternative de novo strategy, P-clouds, that instead searches for clusters of high-abundance oligonucleotides that are related in sequence space (oligo "clouds"). We show here that P-clouds predicts >840 Mbp of additional repetitive sequences in the human genome, thus suggesting that 66%-69% of the human genome is repetitive or repeat-derived. To investigate this remarkable difference, we conducted detailed analyses of the ability of both P-clouds and a commonly used conventional approach, RepeatMasker (RM), to detect different sized fragments of the highly abundant human Alu and MIR SINEs. RM can have surprisingly low sensitivity for even moderately long fragments, in contrast to P-clouds, which has good sensitivity down to small fragment sizes (∼25 bp). Although short fragments have a high intrinsic probability of being false positives, we performed a probabilistic annotation that reflects this fact. We further developed "element-specific" P-clouds (ESPs) to identify novel Alu and MIR SINE elements, and using it we identified ∼100 Mb of previously unannotated human elements. ESP estimates of new MIR sequences are in good agreement with RM-based predictions of the amount that RM missed. These results highlight the need for combined, probabilistic genome annotation approaches and suggest that the human genome consists of substantially more repetitive sequence than previously believed.
转座元件 (TEs) 通常通过与保守元件序列比对来在真核基因组中鉴定。使用这种方法,大约一半的人类基因组已经被鉴定为 TEs 和低复杂度重复序列。我们最近开发了一种高度敏感的新从头策略 P-clouds,它不是搜索序列空间中相关的高丰度寡核苷酸簇(寡核苷酸“云”)。我们在这里表明,P-clouds 预测了人类基因组中 >840 Mbp 的额外重复序列,因此表明人类基因组的 66%-69%是重复的或源自重复序列。为了研究这种显著差异,我们对 P-clouds 和一种常用的传统方法 RepeatMasker (RM) 检测高度丰富的人类 Alu 和 MIR SINE 不同大小片段的能力进行了详细分析。与 P-clouds 不同,RM 对即使是中等长度的片段也具有惊人的低灵敏度,P-clouds 对小片段大小(约 25 bp)具有良好的灵敏度。虽然短片段具有很高的假阳性内在概率,但我们进行了概率注释,反映了这一事实。我们进一步开发了“元素特异性”P-clouds(ESPs)来识别新的 Alu 和 MIR SINE 元件,并使用它我们鉴定了大约 100 Mb 以前未注释的人类元件。ESP 对新 MIR 序列的估计与 RM 基于 RM 错过的量的预测非常吻合。这些结果强调了需要联合、概率基因组注释方法,并表明人类基因组包含的重复序列比以前认为的要多得多。