Di Blasi Claudia, Bellafiore Emanuela, Salih Mustafa Am, Manzini M Chiara, Moore Steven A, Seidahmed Mohammed Z, Mukhtar Maowia M, Karrar Zein A, Walsh Christopher A, Campbell Kevin P, Mantegazza Renato, Morandi Lucia, Mora Marina
Division of Neuromuscular Diseases and Neuroimmunology, Fondazione IRCCS Istituto Neurologico C, Besta, Milan, Italy.
BMC Res Notes. 2011 Dec 13;4:534. doi: 10.1186/1756-0500-4-534.
Congenital muscular dystrophy type 1A is caused by mutations in the LAMA2 gene that encodes the laminin α2 chain, a component of the skeletal muscle extracellular matrix protein laminin-211. The clinical spectrum of the disease is more heterogeneous than previously thought, particularly in terms of motor achievement and disease progression. We investigated clinical findings and performed molecular genetic analysis in 3 families from Saudi Arabia and 1 from Sudan in whom congenital muscular dystrophy 1A was suspected based on homozygosity mapping and laminin α2 chain deficiency.
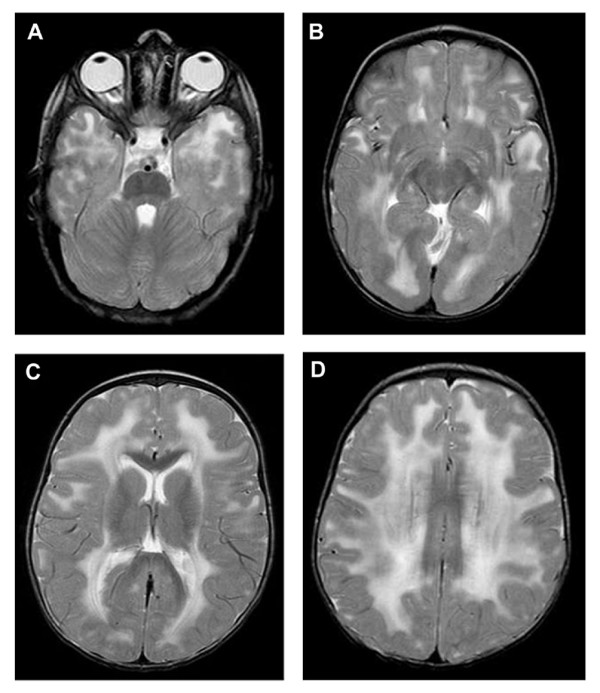
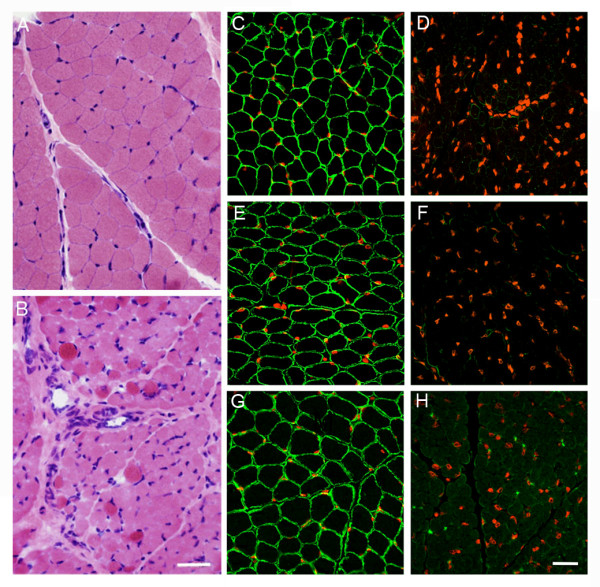
We investigated 9 affected individuals from 1 Sudanese and 3 Saudi families in whom MDC1A was suggested by clinical, neuroimaging and/or pathological findings and by homozygosity mapping at the LAMA2 locus. Morphological and immunohistochemical analysis were performed in 3 patients from the 3 Saudi families. SSCP analysis, DNA sequencing and microsatellite analysis were carried out in the 4 index cases.
A previously described mutation in the LAMA2 gene, a homozygous T > C substitution at position +2 of the consensus donor splice site of exon 26, was found in the 4 index patients. Clinical evaluation of 9 patients from the 4 families revealed variable disease severity particularly as regards motor achievement and disease progression. Microsatellite analysis showed an identical mutation-associated haplotype in the 4 index cases indicating a founder effect of the mutation in all 4 families.
Our data provide further evidence that the clinical spectrum of MDC1A due to a single mutation is heterogeneous, particularly in terms of motor achievement and disease progression, making it difficult to give a reliable prognosis even in patients with identical LAMA2-associated haplotype. The c.3924 + 2 T > C mutation to date has been found only in patients originating from the Middle East or Sudan; therefore laminin 2 chain deficiency in patients from those regions should initially prompt a search for this mutation.
1A型先天性肌营养不良是由编码层粘连蛋白α2链的LAMA2基因突变引起的,层粘连蛋白α2链是骨骼肌细胞外基质蛋白层粘连蛋白-211的一个组成部分。该疾病的临床谱比之前认为的更加异质性,尤其是在运动能力和疾病进展方面。我们对来自沙特阿拉伯的3个家庭和来自苏丹的1个家庭进行了临床研究并开展了分子遗传学分析,这些家庭基于纯合子定位和层粘连蛋白α2链缺乏被怀疑患有1A型先天性肌营养不良。
我们调查了来自1个苏丹家庭和3个沙特家庭的9名受影响个体,这些个体根据临床、神经影像学和/或病理学发现以及LAMA2基因座的纯合子定位提示患有MDC1A。对来自3个沙特家庭的3名患者进行了形态学和免疫组织化学分析。对4例先证者进行了单链构象多态性分析、DNA测序和微卫星分析。
在4例先证患者中发现了LAMA2基因中一个先前描述的突变,即外显子26共有供体剪接位点+2位置的纯合T>C替换。对来自这4个家庭的9名患者的临床评估显示疾病严重程度各异,尤其是在运动能力和疾病进展方面。微卫星分析显示4例先证者中存在相同的与突变相关的单倍型,表明该突变在所有4个家庭中存在奠基者效应。
我们的数据进一步证明,由单一突变导致的MDC1A临床谱是异质性的,尤其是在运动能力和疾病进展方面,这使得即使是具有相同LAMA2相关单倍型的患者也难以给出可靠的预后。迄今为止,c.3924+2T>C突变仅在来自中东或苏丹的患者中发现;因此,来自这些地区的患者出现层粘连蛋白2链缺乏时应首先排查该突变。