Faculty of Medicine, Discipline of Medicine and Genetics, Memorial University, St. John's, Newfoundland, Canada.
PLoS One. 2011;6(12):e28853. doi: 10.1371/journal.pone.0028853. Epub 2011 Dec 14.
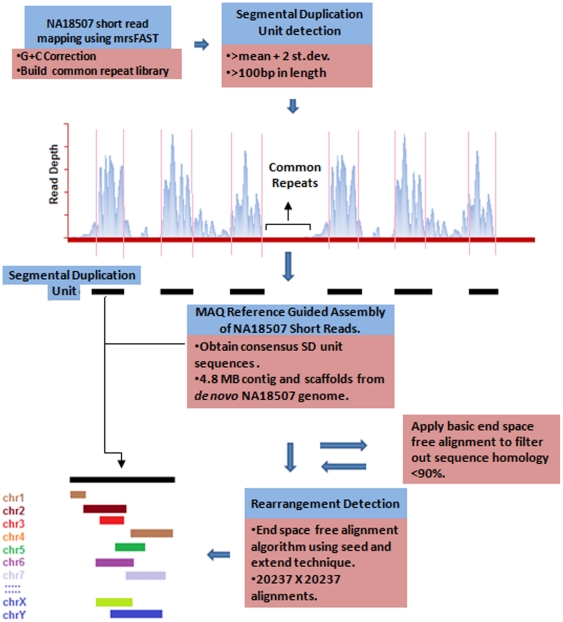
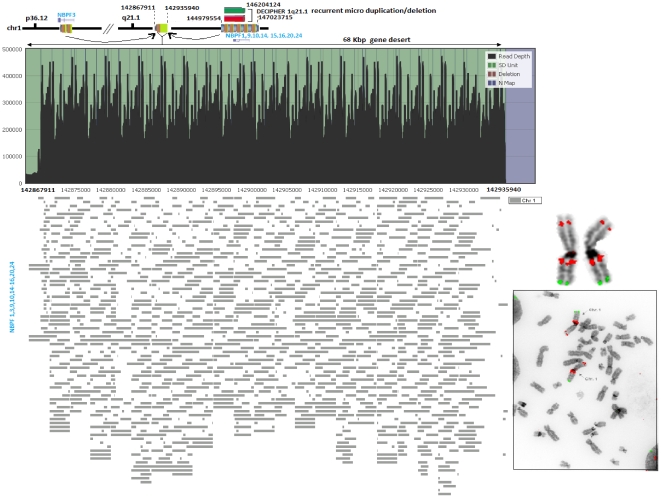
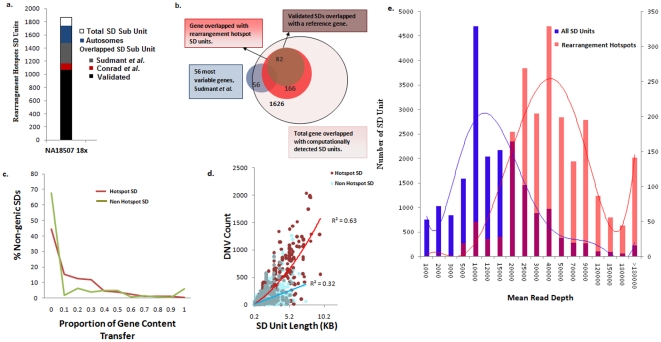
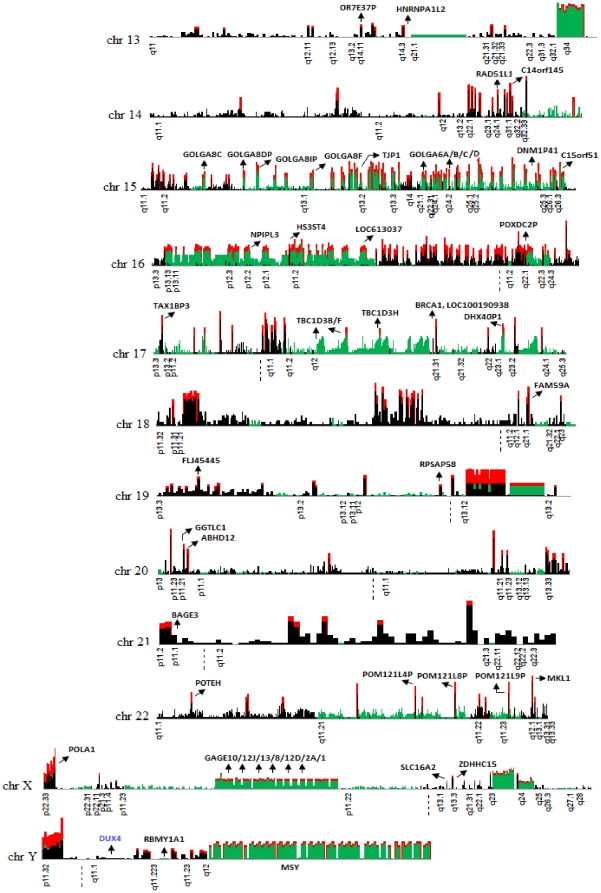
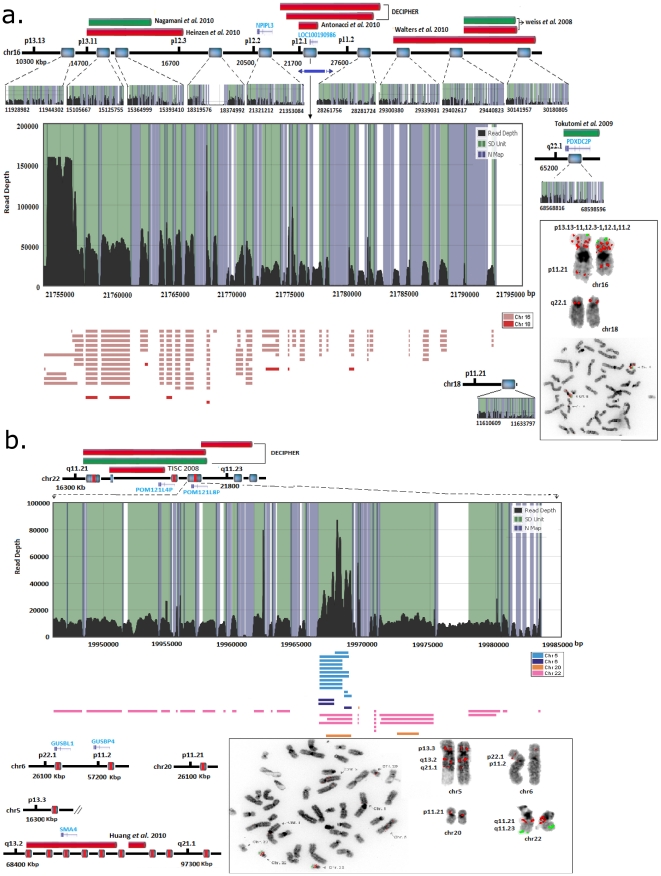
The primary objective of this study was to create a genome-wide high resolution map (i.e., >100 bp) of 'rearrangement hotspots' which can facilitate the identification of regions capable of mediating de novo deletions or duplications in humans. A hierarchical method was employed to fragment segmental duplications (SDs) into multiple smaller SD units. Combining an end space free pairwise alignment algorithm with a 'seed and extend' approach, we have exhaustively searched 409 million alignments to detect complex structural rearrangements within the reference-guided assembly of the NA18507 human genome (18× coverage), including the previously identified novel 4.8 Mb sequence from de novo assembly within this genome. We have identified 1,963 rearrangement hotspots within SDs which encompass 166 genes and display an enrichment of duplicated gene nucleotide variants (DNVs). These regions are correlated with increased non-allelic homologous recombination (NAHR) event frequency which presumably represents the origin of copy number variations (CNVs) and pathogenic duplications/deletions. Analysis revealed that 20% of the detected hotspots are clustered within the proximal and distal SD breakpoints flanked by the pathogenic deletions/duplications that have been mapped for 24 NAHR-mediated genomic disorders. FISH Validation of selected complex regions revealed 94% concordance with in silico localization of the highly homologous derivatives. Other results from this study indicate that intra-chromosomal recombination is enhanced in genic compared with agenic duplicated regions, and that gene desert regions comprising SDs may represent reservoirs for creation of novel genes. The generation of genome-wide signatures of 'rearrangement hotspots', which likely serve as templates for NAHR, may provide a powerful approach towards understanding the underlying mutational mechanism(s) for development of constitutional and acquired diseases.
本研究的主要目的是创建一个全基因组高分辨率图谱(即>100bp),以识别能够介导人类新生缺失或重复的区域。我们采用了一种分层方法将串联重复序列(SD)分割成多个较小的 SD 单元。通过将一个无末端间隙的两两比对算法与“种子和扩展”方法相结合,我们对 4.09 亿个比对进行了全面搜索,以检测人类参考基因组 NA18507 (18×覆盖度)引导组装中的复杂结构重排,包括先前在该基因组从头组装中鉴定出的新的 4.8Mb 序列。我们在 SD 中鉴定出了 1963 个重排热点,这些热点包含 166 个基因,并显示出重复基因核苷酸变体(DNV)的富集。这些区域与增加的非等位基因同源重组(NAHR)事件频率相关,这可能代表了拷贝数变异(CNV)和致病性重复/缺失的起源。分析表明,检测到的热点中有 20%聚类在致病性缺失/重复的近端和远端 SD 断点内,这些致病性缺失/重复已被映射到 24 种 NAHR 介导的基因组疾病中。对选定的复杂区域进行的 FISH 验证显示,与高同源衍生的计算机定位有 94%的一致性。本研究的其他结果表明,与非基因区相比,基因内的重组在基因重复区更为增强,并且包含 SD 的基因荒漠区可能是新基因产生的库。全基因组“重排热点”特征的产生,可能作为 NAHR 的模板,为理解先天和后天疾病的潜在突变机制提供了一种强大的方法。