St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, New York, United States of America.
PLoS One. 2012;7(1):e29708. doi: 10.1371/journal.pone.0029708. Epub 2012 Jan 4.
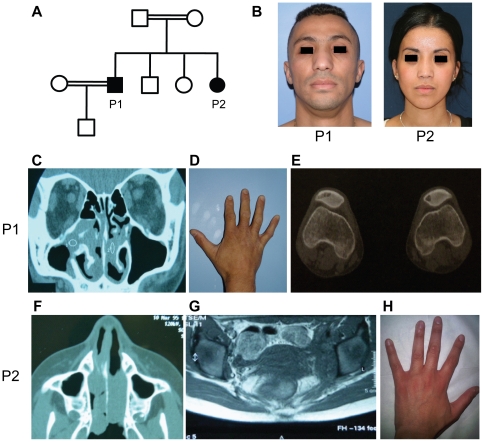
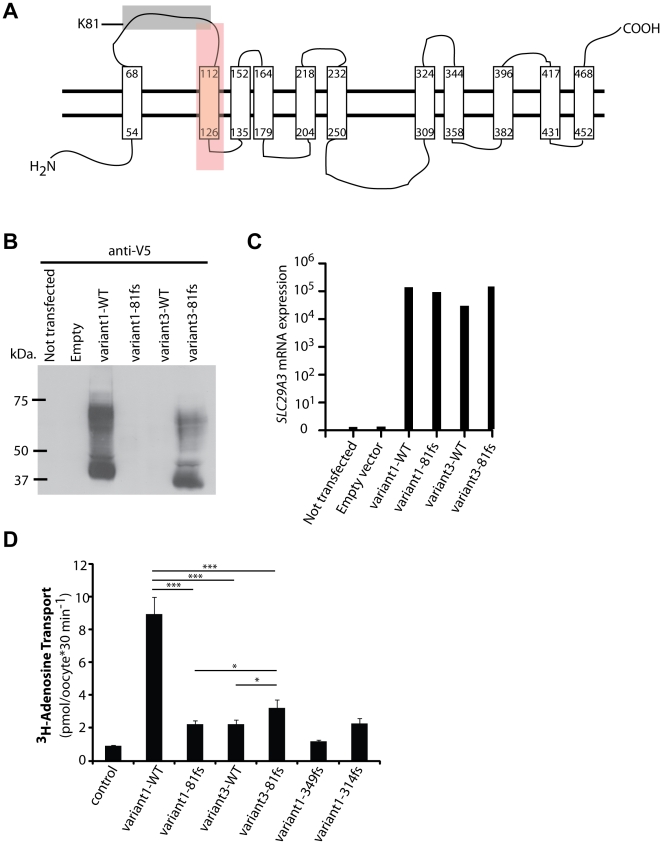
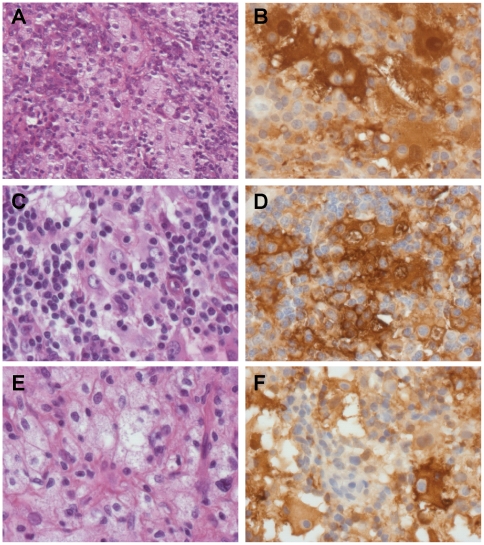
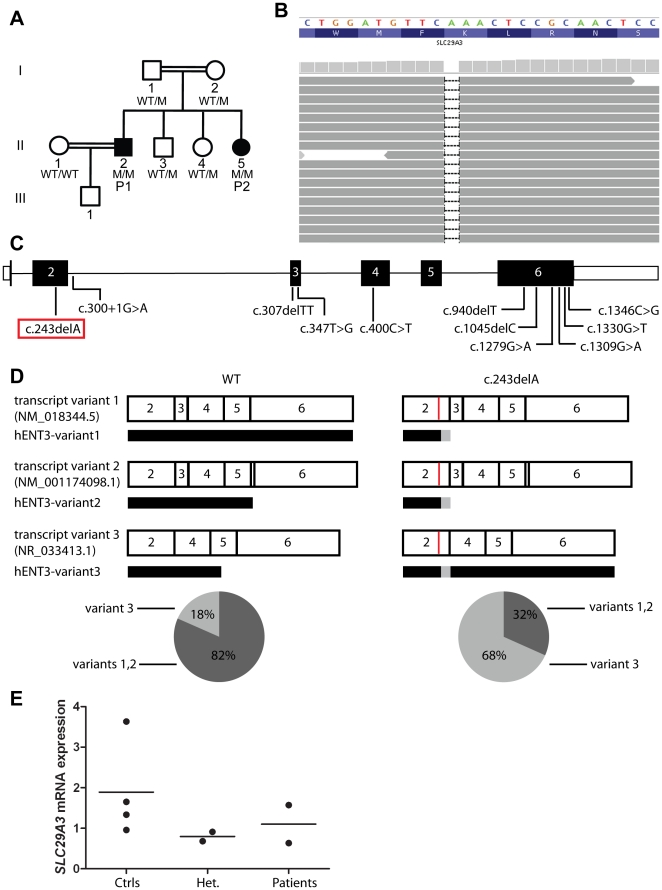
We investigated two siblings with granulomatous histiocytosis prominent in the nasal area, mimicking rhinoscleroma and Rosai-Dorfman syndrome. Genome-wide linkage analysis and whole-exome sequencing identified a homozygous frameshift deletion in SLC29A3, which encodes human equilibrative nucleoside transporter-3 (hENT3). Germline mutations in SLC29A3 have been reported in rare patients with a wide range of overlapping clinical features and inherited disorders including H syndrome, pigmented hypertrichosis with insulin-dependent diabetes, and Faisalabad histiocytosis. With the exception of insulin-dependent diabetes and mild finger and toe contractures in one sibling, the two patients with nasal granulomatous histiocytosis studied here displayed none of the many SLC29A3-associated phenotypes. This mild clinical phenotype probably results from a remarkable genetic mechanism. The SLC29A3 frameshift deletion prevents the expression of the normally coding transcripts. It instead leads to the translation, expression, and function of an otherwise noncoding, out-of-frame mRNA splice variant lacking exon 3 that is eliminated by nonsense-mediated mRNA decay (NMD) in healthy individuals. The mutated isoform differs from the wild-type hENT3 by the modification of 20 residues in exon 2 and the removal of another 28 amino acids in exon 3, which include the second transmembrane domain. As a result, this new isoform displays some functional activity. This mechanism probably accounts for the narrow and mild clinical phenotype of the patients. This study highlights the 'rescue' role played by a normally noncoding mRNA splice variant of SLC29A3, uncovering a new mechanism by which frameshift mutations can be hypomorphic.
我们研究了两名在鼻腔区域表现突出的肉芽肿性组织细胞增多症的兄弟姐妹,其表现类似于类肉瘤和罗萨达-多夫曼综合征。全基因组连锁分析和全外显子组测序鉴定出 SLC29A3 的纯合移码缺失,该基因编码人类平衡核苷转运蛋白 3(hENT3)。SLC29A3 的种系突变已在罕见的具有广泛重叠临床表现和遗传疾病的患者中报道,包括 H 综合征、伴胰岛素依赖性糖尿病的色素性多毛症和费萨拉巴德组织细胞增多症。除了一名兄弟姐妹存在胰岛素依赖性糖尿病和轻微的手指和脚趾挛缩外,这里研究的两名鼻腔肉芽肿性组织细胞增多症患者没有表现出 SLC29A3 相关表型的任何一种。这种轻度临床表型可能是由于一种显著的遗传机制。SLC29A3 的移码缺失阻止了正常编码转录物的表达。相反,它导致外显子 3 缺失的无意义 mRNA 降解(NMD)在健康个体中缺失的非编码、移码 mRNA 剪接变体的翻译、表达和功能。突变型同工型与野生型 hENT3 的不同之处在于外显子 2 中的 20 个残基的修饰和外显子 3 中另 28 个氨基酸的缺失,其中包括第二个跨膜域。因此,这种新的同工型具有一些功能活性。这种机制可能解释了患者的狭窄和轻度临床表型。本研究强调了 SLC29A3 的正常非编码 mRNA 剪接变体的“拯救”作用,揭示了一种新的机制,通过该机制,移码突变可以表现为低功能。