Wellchild Paediatric Research Centre and Department of Medical and Molecular Genetics, University of Birmingham College of Medical and Dental Sciences, Edgbaston, Birmingham, United Kingdom.
PLoS Genet. 2010 Feb 5;6(2):e1000833. doi: 10.1371/journal.pgen.1000833.
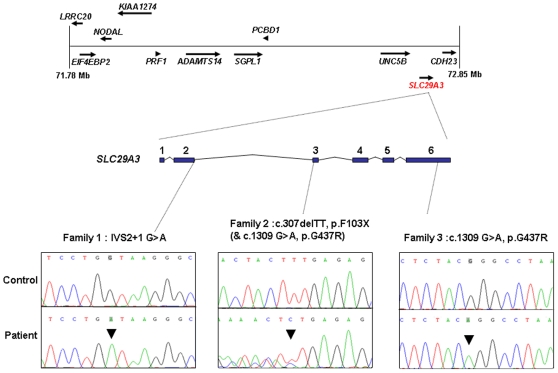
The histiocytoses are a heterogeneous group of disorders characterised by an excessive number of histiocytes. In most cases the pathophysiology is unclear and treatment is nonspecific. Faisalabad histiocytosis (FHC) (MIM 602782) has been classed as an autosomal recessively inherited form of histiocytosis with similarities to Rosai-Dorfman disease (RDD) (also known as sinus histiocytosis with massive lymphadenopathy (SHML)). To elucidate the molecular basis of FHC, we performed autozygosity mapping studies in a large consanguineous family and identified a novel locus at chromosome 10q22.1. Mutation analysis of candidate genes within the target interval identified biallelic germline mutations in SLC29A3 in the FHC kindred and in two families reported to have familial RDD. Analysis of SLC29A3 expression during mouse embryogenesis revealed widespread expression by e14.5 with prominent expression in the central nervous system, eye, inner ear, and epithelial tissues including the gastrointestinal tract. SLC29A3 encodes an intracellular equilibrative nucleoside transporter (hENT3) with affinity for adenosine. Recently germline mutations in SLC29A3 were also described in two rare autosomal recessive disorders with overlapping phenotypes: (a) H syndrome (MIM 612391) that is characterised by cutaneous hyperpigmentation and hypertrichosis, hepatomegaly, heart anomalies, hearing loss, and hypogonadism; and (b) PHID (pigmented hypertrichosis with insulin-dependent diabetes mellitus) syndrome. Our findings suggest that a variety of clinical diagnoses (H and PHID syndromes, FHC, and familial RDD) can be included in a new diagnostic category of SLC29A3 spectrum disorder.
组织细胞增多症是一组以组织细胞数量过多为特征的异质性疾病。在大多数情况下,其病理生理学机制尚不清楚,且治疗方法不具特异性。费萨拉巴德组织细胞增多症(FHC)(MIM 602782)被归类为一种常染色体隐性遗传形式的组织细胞增多症,与罗萨-多夫曼病(RDD)(也称为伴有巨大淋巴结病的窦组织细胞增生症(SHML))具有相似性。为了阐明 FHC 的分子基础,我们对一个大型近亲家族进行了自交系作图研究,在 10q22.1 染色体上确定了一个新的位点。对目标区间内候选基因的突变分析在 FHC 家系中发现了 SLC29A3 的双等位基因种系突变,在两个报道有家族性 RDD 的家系中也发现了 SLC29A3 的双等位基因种系突变。在小鼠胚胎发生过程中对 SLC29A3 表达的分析显示,e14.5 时广泛表达,在中枢神经系统、眼睛、内耳和包括胃肠道在内的上皮组织中表达明显。SLC29A3 编码一种细胞内核苷转运蛋白(hENT3),对腺苷具有亲和力。最近,SLC29A3 的种系突变也在两种具有重叠表型的罕见常染色体隐性疾病中被描述:(a)H 综合征(MIM 612391),其特征为皮肤色素沉着过度和多毛症、肝肿大、心脏异常、听力损失和性腺功能减退;和(b)PHID(色素性多毛症伴胰岛素依赖型糖尿病)综合征。我们的发现表明,各种临床诊断(H 和 PHID 综合征、FHC 和家族性 RDD)可以被归入一个新的 SLC29A3 谱障碍的诊断类别。