Department of Proteomics and Signal Transduction, Max-Planck Institute of Biochemistry, Am Klopferspitz 18, D-82152 Martinsried, Germany.
Mol Cell Proteomics. 2012 Mar;11(3):M111.014050. doi: 10.1074/mcp.M111.014050. Epub 2012 Jan 25.
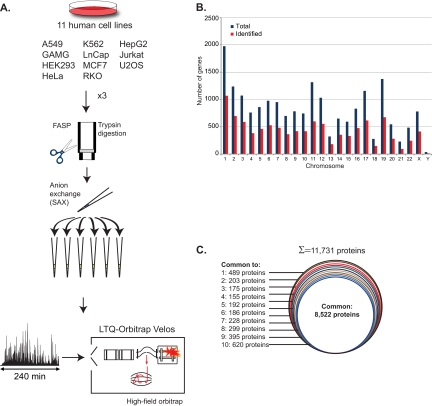
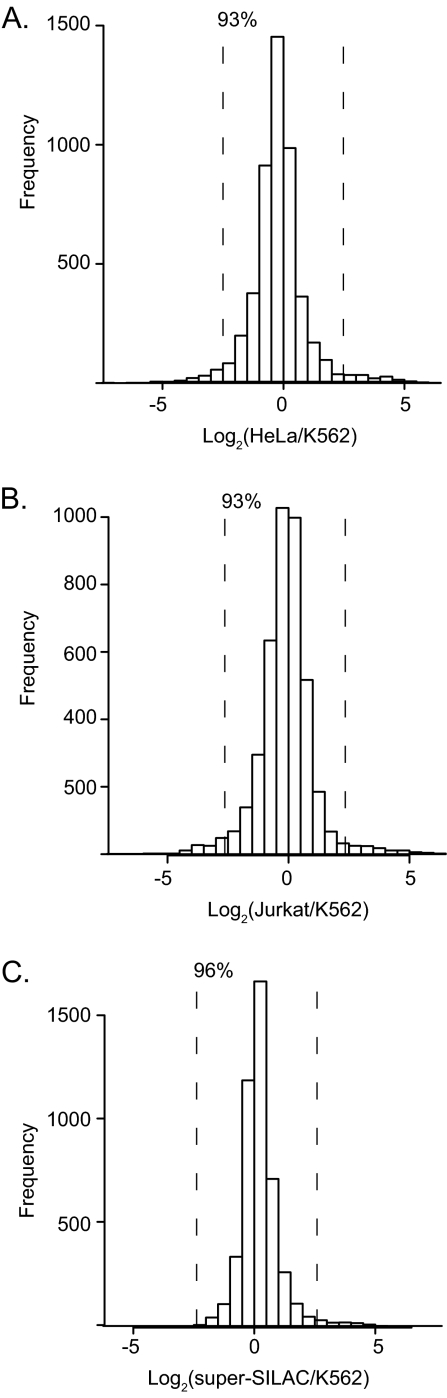
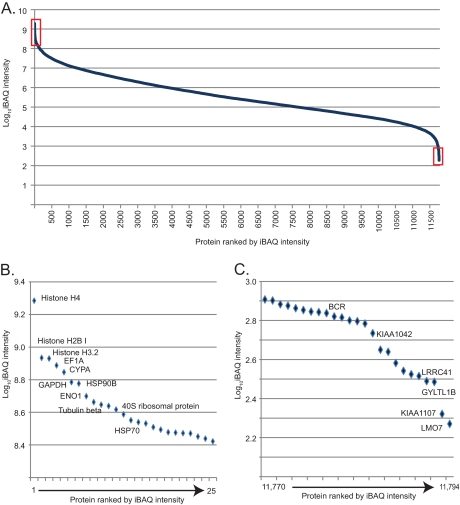
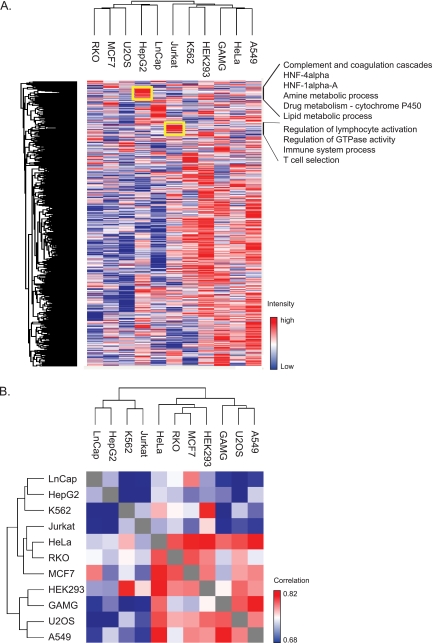
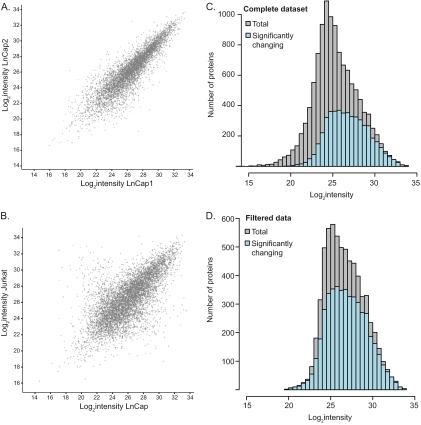
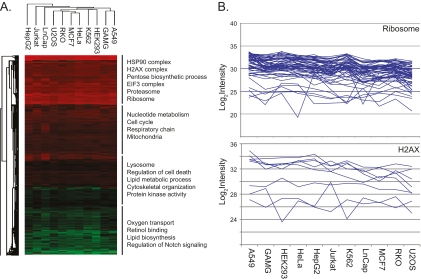
Deep proteomic analysis of mammalian cell lines would yield an inventory of the building blocks of the most commonly used systems in biological research. Mass spectrometry-based proteomics can identify and quantify proteins in a global and unbiased manner and can highlight the cellular processes that are altered between such systems. We analyzed 11 human cell lines using an LTQ-Orbitrap family mass spectrometer with a "high field" Orbitrap mass analyzer with improved resolution and sequencing speed. We identified a total of 11,731 proteins, and on average 10,361 ± 120 proteins in each cell line. This very high proteome coverage enabled analysis of a broad range of processes and functions. Despite the distinct origins of the cell lines, our quantitative results showed surprisingly high similarity in terms of expressed proteins. Nevertheless, this global similarity of the proteomes did not imply equal expression levels of individual proteins across the 11 cell lines, as we found significant differences in expression levels for an estimated two-third of them. The variability in cellular expression levels was similar for low and high abundance proteins, and even many of the most highly expressed proteins with household roles showed significant differences between cells. Metabolic pathways, which have high redundancy, exhibited variable expression, whereas basic cellular functions such as the basal transcription machinery varied much less. We harness knowledge of these cell line proteomes for the construction of a broad coverage "super-SILAC" quantification standard. Together with the accompanying paper (Schaab, C. MCP 2012, PMID: 22301388) (17) these data can be used to obtain reference expression profiles for proteins of interest both within and across cell line proteomes.
哺乳动物细胞系的深度蛋白质组分析将产生生物研究中最常用系统的构建模块清单。基于质谱的蛋白质组学可以以全局和无偏的方式鉴定和定量蛋白质,并可以突出细胞过程之间的改变。我们使用 LTQ-Orbitrap 系列质谱仪和具有改进分辨率和测序速度的“高场”Orbitrap 质量分析仪分析了 11 个人类细胞系。我们总共鉴定了 11731 种蛋白质,平均每种细胞系鉴定到 10361 ± 120 种蛋白质。这种非常高的蛋白质组覆盖率使我们能够分析广泛的过程和功能。尽管细胞系的起源不同,但我们的定量结果显示,表达的蛋白质在很大程度上具有惊人的相似性。然而,这种蛋白质组的全局相似性并不意味着个体蛋白质在 11 种细胞系中的表达水平相等,因为我们发现它们中的约三分之二的蛋白质表达水平存在显著差异。低丰度和高丰度蛋白质的细胞表达水平变化相似,甚至许多具有家庭作用的高度表达蛋白质在细胞之间也表现出显著差异。具有高冗余性的代谢途径表现出可变的表达,而基本的细胞功能,如基础转录机制,变化则要小得多。我们利用这些细胞系蛋白质组学的知识构建了一个广泛覆盖的“超级 SILAC”定量标准。结合随附的论文(Schaab,C. MCP 2012,PMID:22301388)(17),这些数据可用于获得感兴趣的蛋白质在细胞系蛋白质组内和跨细胞系蛋白质组的参考表达谱。