Department of Statistics, Brigham Young University, Provo, Utah, United States of America.
PLoS One. 2012;7(2):e30996. doi: 10.1371/journal.pone.0030996. Epub 2012 Feb 15.
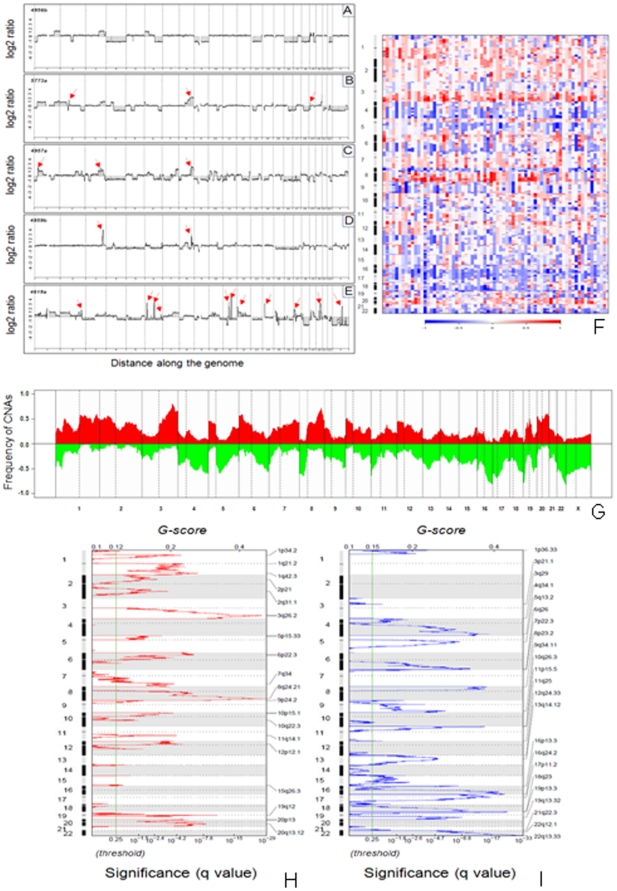
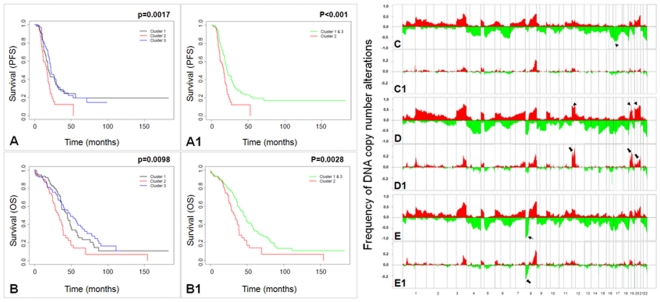
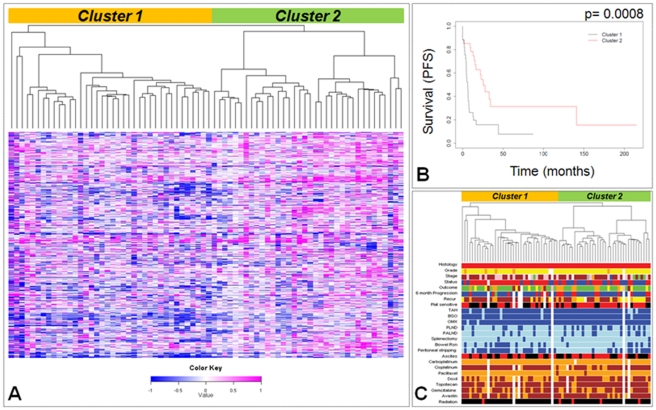
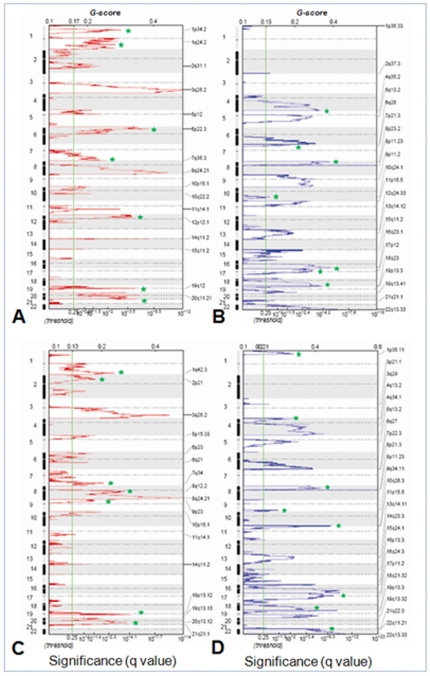
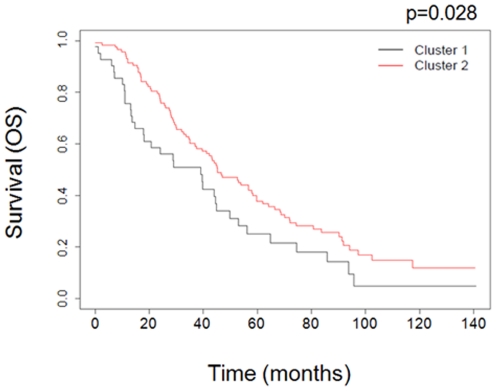
Ovarian cancer is the fifth leading cause of cancer death in women. Ovarian cancers display a high degree of complex genetic alterations involving many oncogenes and tumor suppressor genes. Analysis of the association between genetic alterations and clinical endpoints such as survival will lead to improved patient management via genetic stratification of patients into clinically relevant subgroups. In this study, we aim to define subgroups of high-grade serous ovarian carcinomas that differ with respect to prognosis and overall survival. Genome-wide DNA copy number alterations (CNAs) were measured in 72 clinically annotated, high-grade serous tumors using high-resolution oligonucleotide arrays. Two clinically annotated, independent cohorts were used for validation. Unsupervised hierarchical clustering of copy number data derived from the 72 patient cohort resulted in two clusters with significant difference in progression free survival (PFS) and a marginal difference in overall survival (OS). GISTIC analysis of the two clusters identified altered regions unique to each cluster. Supervised clustering of two independent large cohorts of high-grade serous tumors using the classification scheme derived from the two initial clusters validated our results and identified 8 genomic regions that are distinctly different among the subgroups. These 8 regions map to 8p21.3, 8p23.2, 12p12.1, 17p11.2, 17p12, 19q12, 20q11.21 and 20q13.12; and harbor potential oncogenes and tumor suppressor genes that are likely to be involved in the pathogenesis of ovarian carcinoma. We have identified a set of genetic alterations that could be used for stratification of high-grade serous tumors into clinically relevant treatment subgroups.
卵巢癌是女性癌症死亡的第五大主要原因。卵巢癌表现出高度复杂的遗传改变,涉及许多癌基因和肿瘤抑制基因。分析遗传改变与生存等临床终点之间的关联,将通过将患者按遗传分层为临床相关亚组,从而改善患者管理。在这项研究中,我们旨在定义高级别浆液性卵巢癌的亚组,这些亚组在预后和总生存方面存在差异。使用高分辨率寡核苷酸阵列测量了 72 个临床注释的高级别浆液性肿瘤的全基因组 DNA 拷贝数改变(CNAs)。使用两个临床注释的独立队列进行验证。对来自 72 名患者队列的拷贝数数据进行无监督层次聚类导致无进展生存期(PFS)有显著差异的两个聚类,以及总生存期(OS)有边缘差异的两个聚类。对两个聚类的 GISTIC 分析确定了每个聚类特有的改变区域。使用从两个初始聚类派生的分类方案对两个独立的高级别浆液性肿瘤大队列进行有监督聚类,验证了我们的结果,并确定了亚组之间存在明显差异的 8 个基因组区域。这 8 个区域映射到 8p21.3、8p23.2、12p12.1、17p11.2、17p12、19q12、20q11.21 和 20q13.12;并包含可能参与卵巢癌发病机制的潜在癌基因和肿瘤抑制基因。我们已经确定了一组遗传改变,可用于将高级别浆液性肿瘤分层为临床相关的治疗亚组。