Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, IN 46202, USA.
BMC Genomics. 2011 Dec 23;12 Suppl 5(Suppl 5):S8. doi: 10.1186/1471-2164-12-S5-S8.
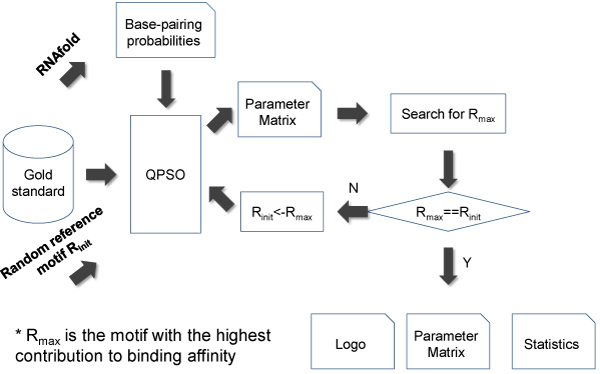
RNA-binding proteins (RBPs) play diverse roles in eukaryotic RNA processing. Despite their pervasive functions in coding and noncoding RNA biogenesis and regulation, elucidating the sequence specificities that define protein-RNA interactions remains a major challenge. Recently, CLIP-seq (Cross-linking immunoprecipitation followed by high-throughput sequencing) has been successfully implemented to study the transcriptome-wide binding patterns of SRSF1, PTBP1, NOVA and fox2 proteins. These studies either adopted traditional methods like Multiple EM for Motif Elicitation (MEME) to discover the sequence consensus of RBP's binding sites or used Z-score statistics to search for the overrepresented nucleotides of a certain size. We argue that most of these methods are not well-suited for RNA motif identification, as they are unable to incorporate the RNA structural context of protein-RNA interactions, which may affect to binding specificity. Here, we describe a novel model-based approach--RNAMotifModeler to identify the consensus of protein-RNA binding regions by integrating sequence features and RNA secondary structures.
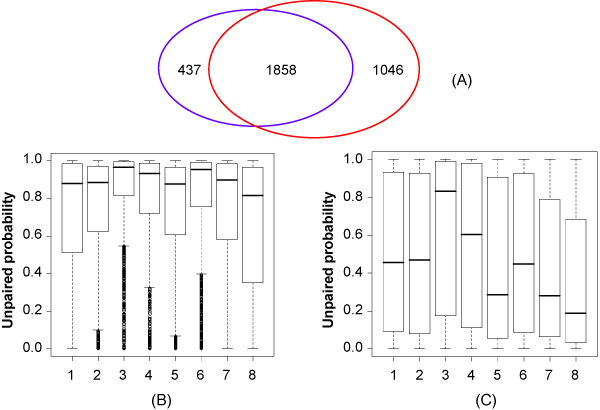
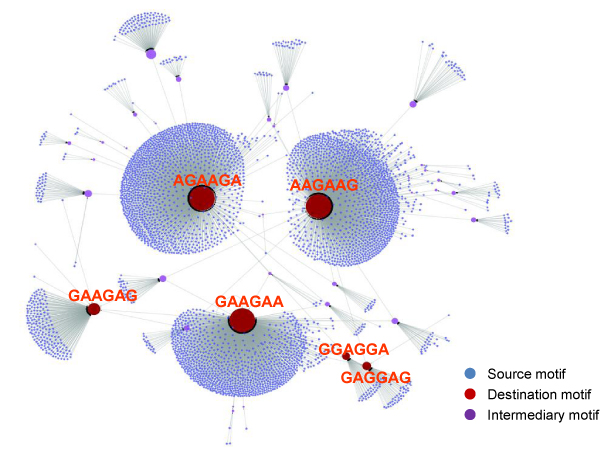
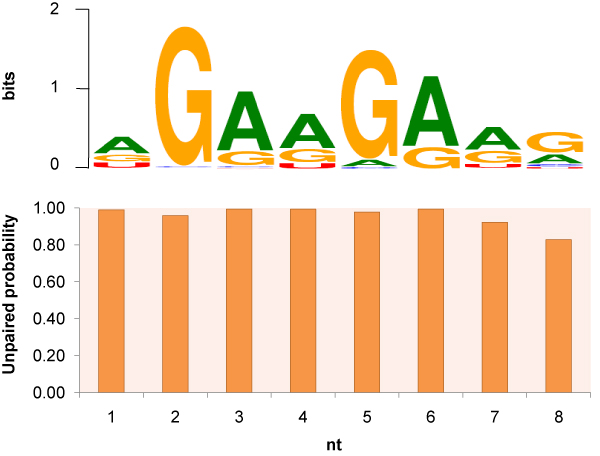
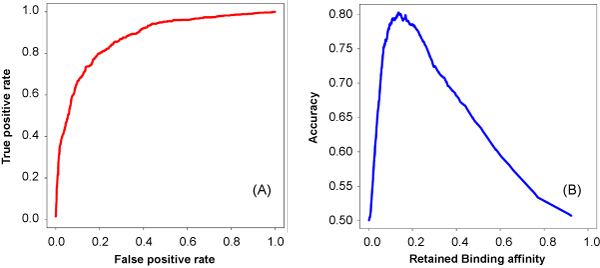
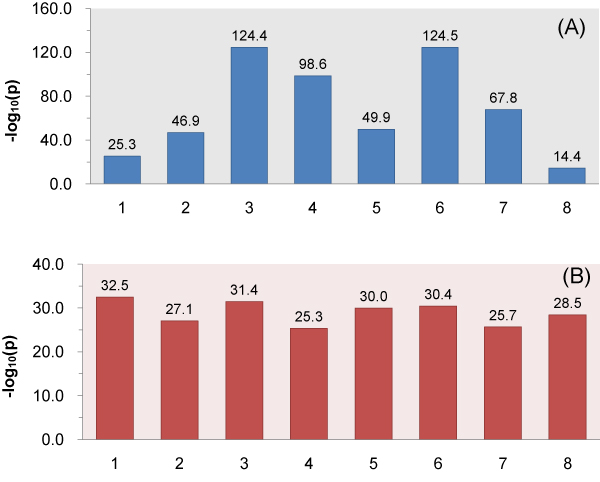
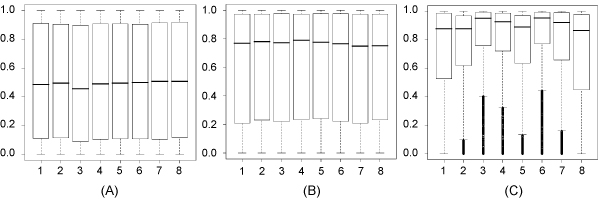
As an example, we implemented RNAMotifModeler on SRSF1 (SF2/ASF) CLIP-seq data. The sequence-structural consensus we identified is a purine-rich octamer 'AGAAGAAG' in a highly single-stranded RNA context. The unpaired probabilities, the probabilities of not forming pairs, are significantly higher than negative controls and the flanking sequence surrounding the binding site, indicating that SRSF1 proteins tend to bind on single-stranded RNA. Further statistical evaluations revealed that the second and fifth bases of SRSF1octamer motif have much stronger sequence specificities, but weaker single-strandedness, while the third, fourth, sixth and seventh bases are far more likely to be single-stranded, but have more degenerate sequence specificities. Therefore, we hypothesize that nucleotide specificity and secondary structure play complementary roles during binding site recognition by SRSF1.
In this study, we presented a computational model to predict the sequence consensus and optimal RNA secondary structure for protein-RNA binding regions. The successful implementation on SRSF1 CLIP-seq data demonstrates great potential to improve our understanding on the binding specificity of RNA binding proteins.
RNA 结合蛋白(RBPs)在真核生物 RNA 加工过程中发挥着多样化的作用。尽管它们在编码和非编码 RNA 的生物发生和调控中具有普遍的功能,但阐明定义蛋白-RNA 相互作用的序列特异性仍然是一个主要挑战。最近,CLIP-seq(交联免疫沉淀 followed by 高通量测序)已成功用于研究 SRSF1、PTBP1、NOVA 和 fox2 蛋白的转录组范围结合模式。这些研究要么采用传统方法,如多模式启发式 for 基序提取(MEME)来发现 RBP 结合位点的序列共识,要么使用 Z 分数统计来搜索特定大小的过代表核苷酸。我们认为,这些方法中的大多数都不适合 RNA 基序识别,因为它们无法将蛋白-RNA 相互作用的 RNA 结构上下文纳入考虑,而这可能会影响结合特异性。在这里,我们描述了一种新的基于模型的方法——RNAMotifModeler,通过整合序列特征和 RNA 二级结构来识别蛋白-RNA 结合区域的共识。
作为一个例子,我们在 SRSF1(SF2/ASF)CLIP-seq 数据上实现了 RNAMotifModeler。我们确定的序列-结构共识是一个富含嘌呤的八聚体'AGAAGAAG',在高度单链 RNA 环境中。未配对的概率,即不形成对的概率,明显高于阴性对照和结合位点周围的侧翼序列,表明 SRSF1 蛋白倾向于结合单链 RNA。进一步的统计评估表明,SRSF1 八聚体基序的第二和第五个碱基具有更强的序列特异性,但较弱的单链性,而第三个、第四个、第六个和第七个碱基更有可能是单链的,但具有更多的简并序列特异性。因此,我们假设核苷酸特异性和二级结构在 SRSF1 结合位点识别过程中发挥互补作用。
在这项研究中,我们提出了一种计算模型来预测蛋白-RNA 结合区域的序列共识和最佳 RNA 二级结构。在 SRSF1 CLIP-seq 数据上的成功实施表明,该模型具有很大的潜力,可以提高我们对 RNA 结合蛋白结合特异性的理解。