Yan Xiting, Li Lun, Lee Joon Sang, Zheng Wei, Ferguson John, Zhao Hongyu
Department of Epidemiology and Public Health, Yale University, New Haven, CT 06520, USA.
BMC Proc. 2011 Nov 29;5 Suppl 9(Suppl 9):S27. doi: 10.1186/1753-6561-5-S9-S27.
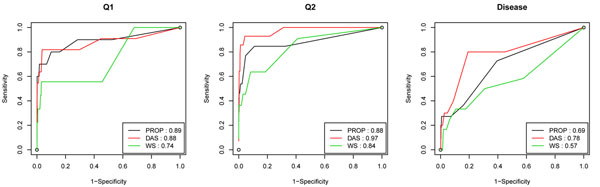
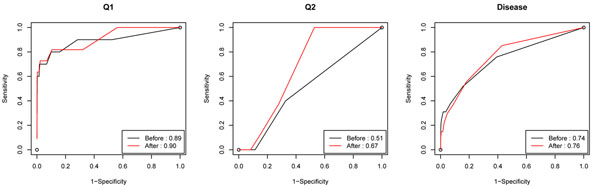
Association studies using tag SNPs have been successful in detecting disease-associated common variants. However, common variants, with rare exceptions, explain only at most 5-10% of the heritability resulting from genetic factors, which leads to the common disease/rare variants assumption. Indeed, recent studies using sequencing technologies have demonstrated that common diseases can be due to rare variants that could not be systematically studied earlier. Unfortunately, methods for common variants are not optimal if applied to rare variants. To identify rare variants that affect disease risk, several investigators have designed new approaches based on the idea of collapsing different rare variants inside the same genomic block (e.g., the same gene or pathway) to enrich the signal. Here, we consider three different collapsing methods in the multimarker regression model and compared their performance on the Genetic Analysis Workshop 17 data using the consistency of results across different simulations and the cross-validation prediction error rate. The comparison shows that the proportion collapsing method seems to outperform the other two methods and can find both truly associated rare and common variants. Moreover, we explore one way of incorporating the functional annotations for the variants in the data that collapses nonsynonymous and synonymous variants separately to allow for different penalties on them. The incorporation of functional annotations led to higher sensitivity and specificity levels when the detection results were compared with the answer sheet. The initial analysis was performed without knowledge of the simulating model.
使用标签单核苷酸多态性(tag SNPs)的关联研究已成功检测出与疾病相关的常见变异。然而,除了极少数例外情况,常见变异最多只能解释遗传因素导致的5%至10%的遗传力,这就引出了常见疾病/罕见变异的假设。事实上,最近使用测序技术的研究表明,常见疾病可能是由早期无法系统研究的罕见变异引起的。不幸的是,如果将适用于常见变异的方法应用于罕见变异,并非最佳选择。为了识别影响疾病风险的罕见变异,一些研究人员基于将同一基因组区域(如同一基因或通路)内的不同罕见变异合并以增强信号的想法,设计了新的方法。在此,我们在多标记回归模型中考虑了三种不同的合并方法,并使用不同模拟结果的一致性和交叉验证预测错误率,在遗传分析研讨会17的数据上比较了它们的性能。比较结果表明,比例合并方法似乎优于其他两种方法,并且能够找到真正相关的罕见和常见变异。此外,我们探索了一种在数据中纳入变异功能注释的方法,该方法分别合并非同义变异和同义变异,以便对它们施加不同的惩罚。与答案表比较检测结果时,纳入功能注释导致更高的敏感性和特异性水平。初始分析是在不了解模拟模型的情况下进行的。