Madsen Bo Eskerod, Browning Sharon R
Bioinformatics Research Center, University of Aarhus, Aarhus C, Denmark.
PLoS Genet. 2009 Feb;5(2):e1000384. doi: 10.1371/journal.pgen.1000384. Epub 2009 Feb 13.
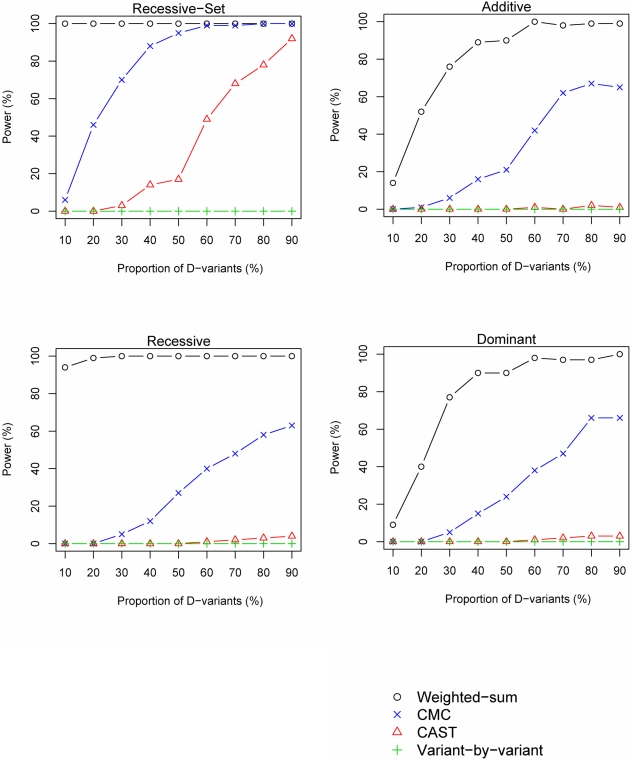
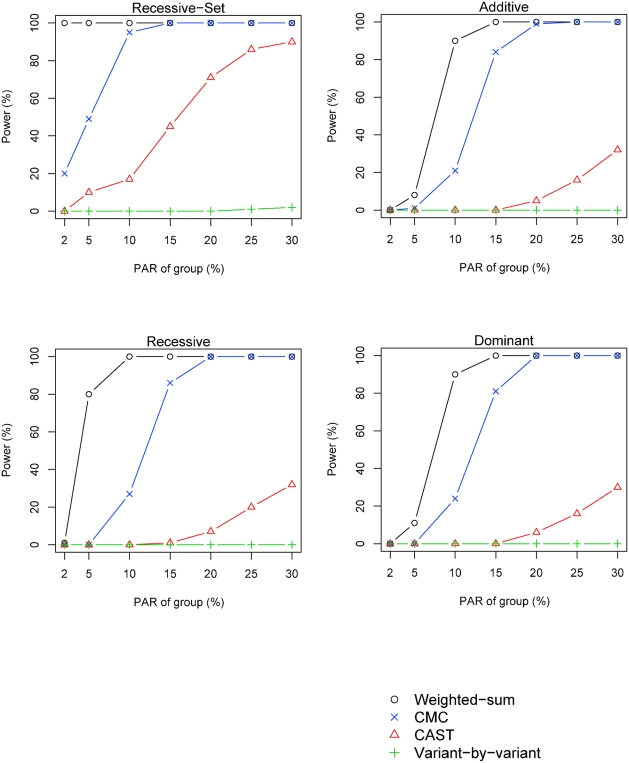
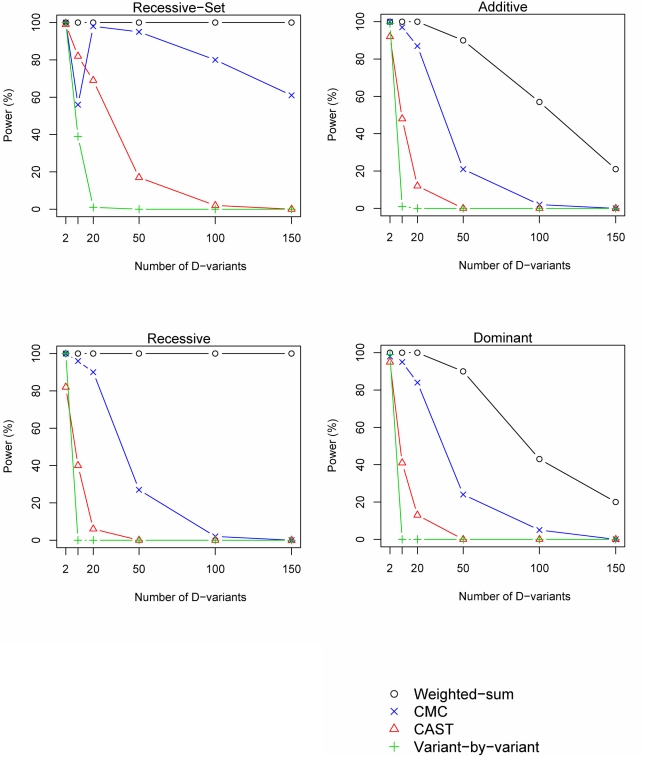
Resequencing is an emerging tool for identification of rare disease-associated mutations. Rare mutations are difficult to tag with SNP genotyping, as genotyping studies are designed to detect common variants. However, studies have shown that genetic heterogeneity is a probable scenario for common diseases, in which multiple rare mutations together explain a large proportion of the genetic basis for the disease. Thus, we propose a weighted-sum method to jointly analyse a group of mutations in order to test for groupwise association with disease status. For example, such a group of mutations may result from resequencing a gene. We compare the proposed weighted-sum method to alternative methods and show that it is powerful for identifying disease-associated genes, both on simulated and Encode data. Using the weighted-sum method, a resequencing study can identify a disease-associated gene with an overall population attributable risk (PAR) of 2%, even when each individual mutation has much lower PAR, using 1,000 to 7,000 affected and unaffected individuals, depending on the underlying genetic model. This study thus demonstrates that resequencing studies can identify important genetic associations, provided that specialised analysis methods, such as the weighted-sum method, are used.
重测序是一种用于识别罕见病相关突变的新兴工具。由于基因分型研究旨在检测常见变异,因此罕见突变很难用单核苷酸多态性(SNP)基因分型来标记。然而,研究表明,遗传异质性可能是常见疾病的一种情况,其中多个罕见突变共同解释了该疾病很大一部分的遗传基础。因此,我们提出了一种加权和方法来联合分析一组突变,以检验其与疾病状态的分组关联。例如,这样一组突变可能来自对一个基因的重测序。我们将提出的加权和方法与其他方法进行比较,结果表明,在模拟数据和ENCODE数据上,该方法在识别疾病相关基因方面都很有效。使用加权和方法,即使每个个体突变的人群归因风险(PAR)低得多,一项重测序研究也能够识别出一个PAR为2%的疾病相关基因,所需的患病人数和未患病人数为1000至7000人,具体数量取决于潜在的遗传模型。因此,这项研究表明,只要使用加权和方法等专门的分析方法,重测序研究就能识别出重要的基因关联。