Zhen Ying, Andolfatto Peter
Department of Ecology and Evolutionary Biology, The Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, NJ, USA.
Methods Mol Biol. 2012;856:141-59. doi: 10.1007/978-1-61779-585-5_6.
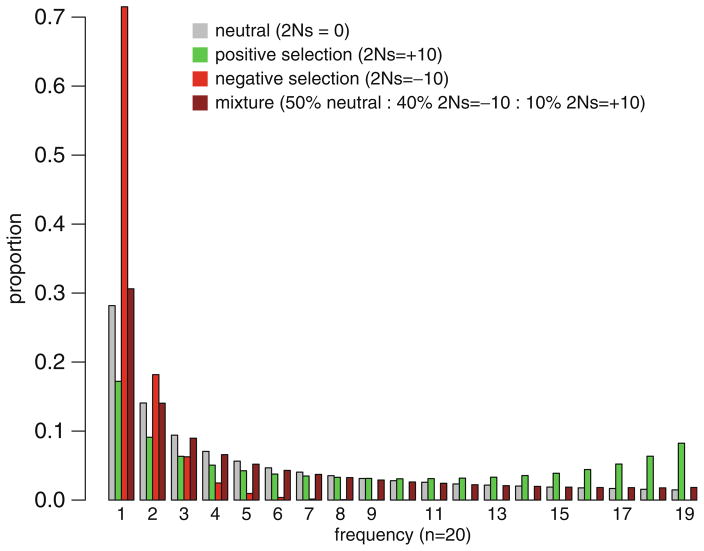
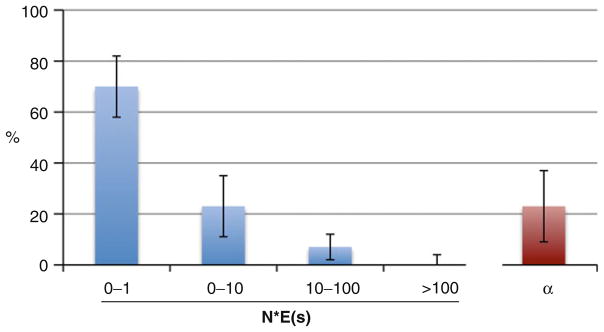
Vast tracts of noncoding DNA contain elements that regulate gene expression in higher eukaryotes. Describing these regulatory elements and understanding how they evolve represent major challenges for biologists. Advances in the ability to survey genome-scale DNA sequence data are providing unprecedented opportunities to use evolutionary models and computational tools to identify functionally important elements and the mode of selection acting on them in multiple species. This chapter reviews some of the current methods that have been developed and applied on noncoding DNA, what they have shown us, and how they are limited. Results of several recent studies reveal that a significantly larger fraction of noncoding DNA in eukaryotic organisms is likely to be functional than previously believed, implying that the functional annotation of most noncoding DNA in these organisms is largely incomplete. In Drosophila, recent studies have further suggested that a large fraction of noncoding DNA divergence observed between species may be the product of recurrent adaptive substitution. Similar studies in humans have revealed a more complex pattern, with signatures of recurrent positive selection being largely concentrated in conserved noncoding DNA elements. Understanding these patterns and the extent to which they generalize to other organisms awaits the analysis of forthcoming genome-scale polymorphism and divergence data from more species.
大片的非编码DNA包含调控高等真核生物基因表达的元件。描述这些调控元件并了解它们如何进化是生物学家面临的主要挑战。在全基因组规模DNA序列数据检测能力方面的进展为利用进化模型和计算工具来识别功能重要元件以及作用于多个物种中这些元件的选择模式提供了前所未有的机会。本章回顾了一些已开发并应用于非编码DNA的当前方法、它们向我们展示了什么以及它们的局限性。最近几项研究的结果表明,真核生物中非编码DNA具有功能的部分可能比以前认为的要大得多,这意味着这些生物中大多数非编码DNA的功能注释在很大程度上是不完整的。在果蝇中,最近的研究进一步表明,物种间观察到的大部分非编码DNA差异可能是反复适应性替代的产物。在人类中的类似研究揭示了一种更复杂的模式,反复正选择的特征主要集中在保守的非编码DNA元件中。要了解这些模式以及它们在多大程度上适用于其他生物,还需要分析来自更多物种的即将出现的全基因组规模多态性和差异数据。