Molecular Pathology and Genetics Division, Kanagawa Cancer Center Research Institute, 1-1-2 Nakao, Asahi-ku, Yokohama 241-0815, Japan.
Nucleic Acids Res. 2012 Jul;40(12):5389-401. doi: 10.1093/nar/gks201. Epub 2012 Mar 8.
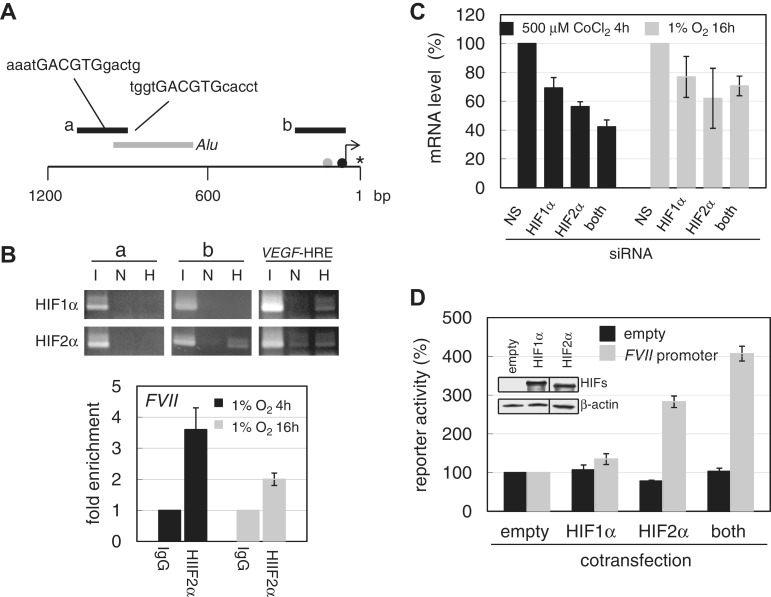
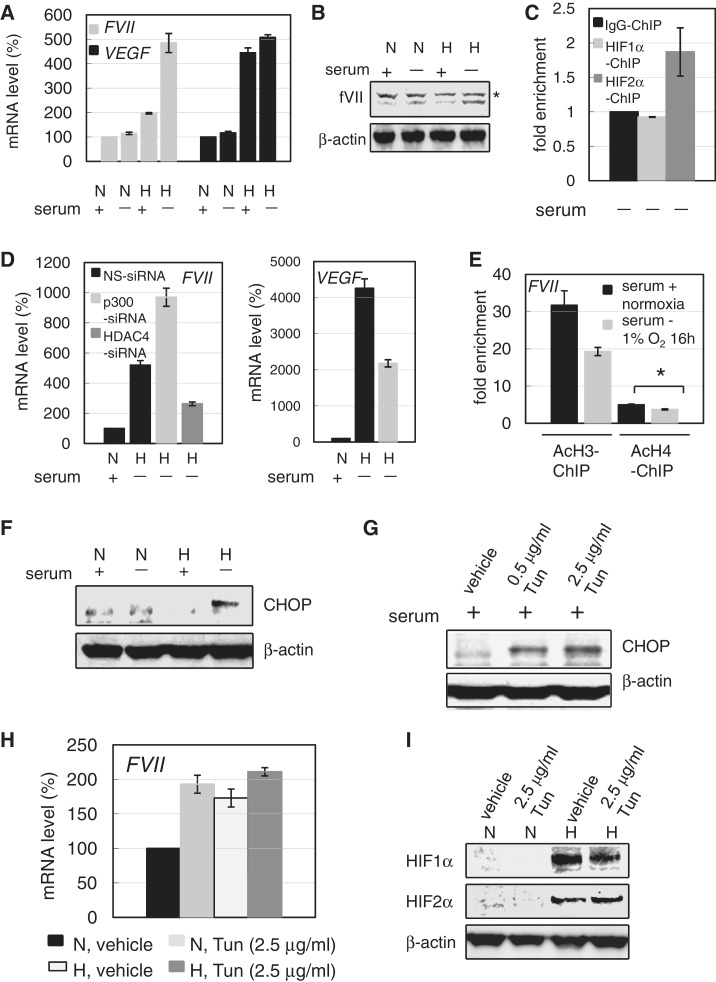
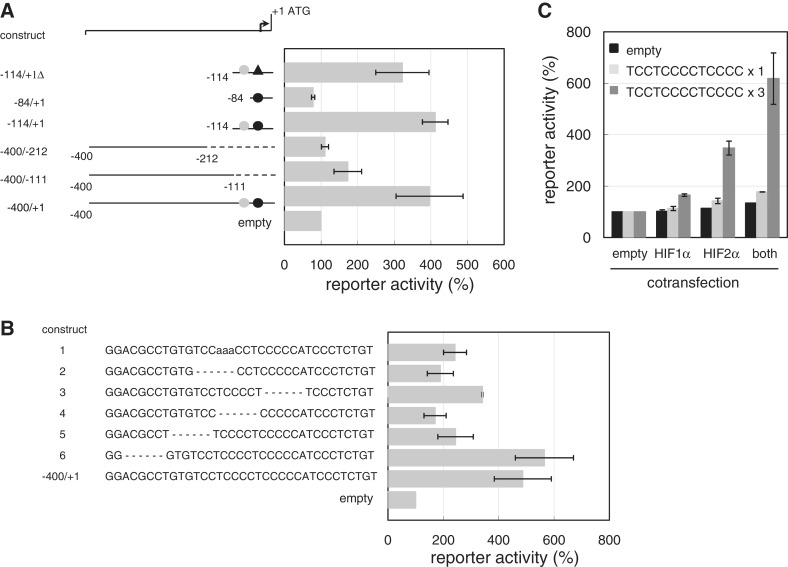
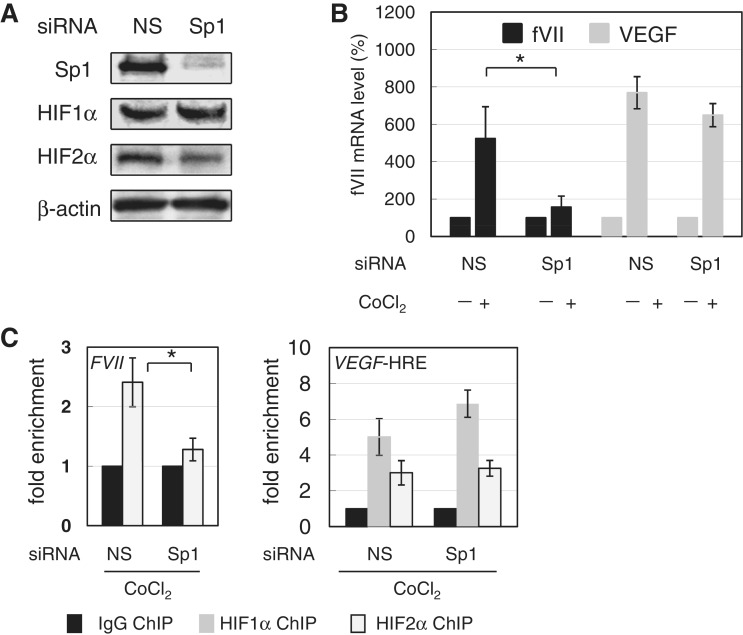
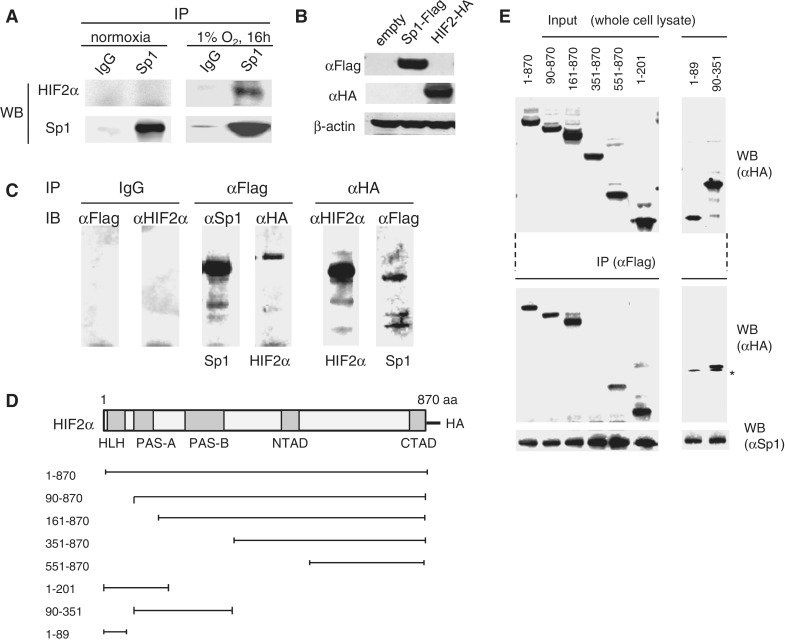
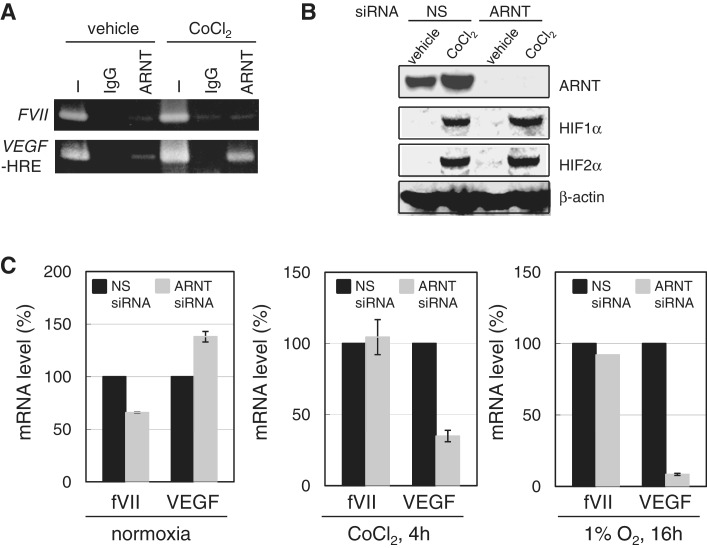
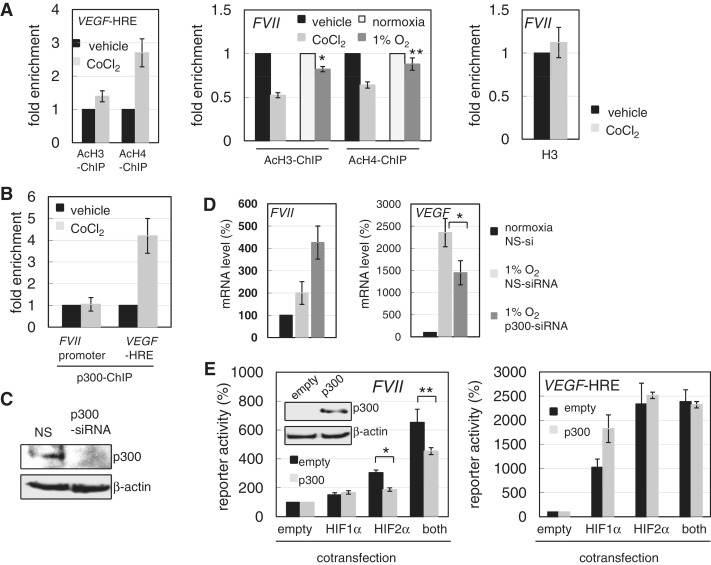
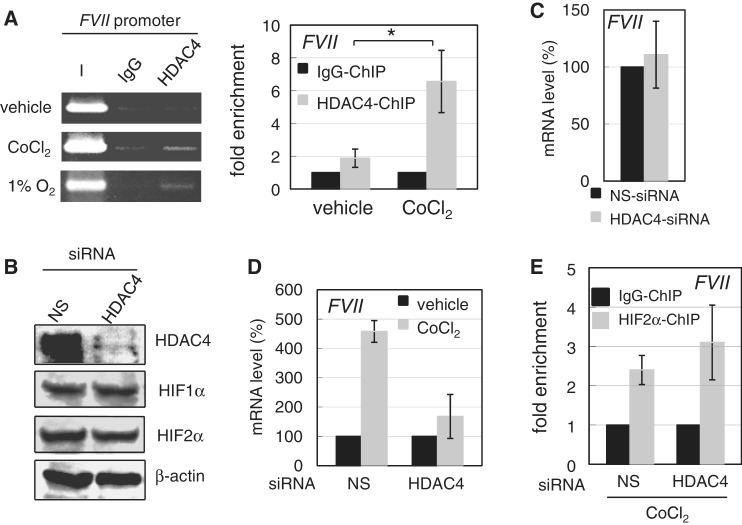
Hypoxia-inducible factors (HIF)-1α and HIF2α are major transcription factors required for adaptive responses to hypoxia. HIFs form a complex with aryl hydrocarbon receptor nuclear translocator (ARNT) to bind to the regulatory regions of target genes. The acetylation of histones by histone acetyltransferases (HATs) is one of the epigenetic marks associated with active chromatin. Indeed, HIFs recruit p300 HAT to hypoxia response elements (HREs) within gene regulatory regions. Here, we report an unusual HIF-mediated transcriptional activation in ovarian clear cell carcinoma (CCC). While characterizing coagulation factor VII (FVII) gene induction during hypoxic conditions, we observed that the interaction of HIF2α with Sp1, but not with ARNT, could induce transcription of FVII in a HRE-independent manner. Unexpectedly, this gene activation is associated with histone deacetylation. We found that a class II HDAC, HDAC4, is recruited with HIF2α to the FVII promoter as a co-activator, while p300 HAT negatively regulated this process. Furthermore, this mechanism can be synergistically enhanced via a deacetylation-dependent pathway when cells are simultaneously exposed to hypoxic and serum-free conditions. These results suggest the presence of a stress-responsive transcription mediated by the HIF2α/Sp1/HDAC4 network and explain how CCC shed their procoagulant activity under hypoxia.
缺氧诱导因子(HIF)-1α和 HIF2α 是适应缺氧反应所必需的主要转录因子。HIF 与芳香烃受体核转位蛋白(ARNT)形成复合物,与靶基因的调节区域结合。组蛋白乙酰转移酶(HATs)对组蛋白的乙酰化是与活性染色质相关的表观遗传标记之一。事实上,HIF 招募 p300 HAT 到基因调节区域内的缺氧反应元件(HRE)。在这里,我们报告了卵巢透明细胞癌(CCC)中一种不寻常的 HIF 介导的转录激活。在研究缺氧条件下凝血因子 VII(FVII)基因诱导时,我们观察到 HIF2α 与 Sp1 的相互作用,而不是与 ARNT 的相互作用,可以以 HRE 非依赖的方式诱导 FVII 的转录。出乎意料的是,这种基因激活与组蛋白去乙酰化有关。我们发现,一种 II 类组蛋白去乙酰化酶(HDAC),即 HDAC4,与 HIF2α 一起被招募到 FVII 启动子上作为共激活子,而 p300 HAT 则负调控这一过程。此外,当细胞同时暴露于缺氧和无血清条件下时,这种机制可以通过去乙酰化依赖性途径协同增强。这些结果表明存在一种由 HIF2α/Sp1/HDAC4 网络介导的应激反应性转录,并解释了 CCC 如何在缺氧下丧失其促凝活性。