Dermatology Service, Veterans Affairs Medical Center, 4150 Clement St, San Francisco, CA 94121, USA.
Clin Dermatol. 2012 May-Jun;30(3):311-22. doi: 10.1016/j.clindermatol.2011.08.017.
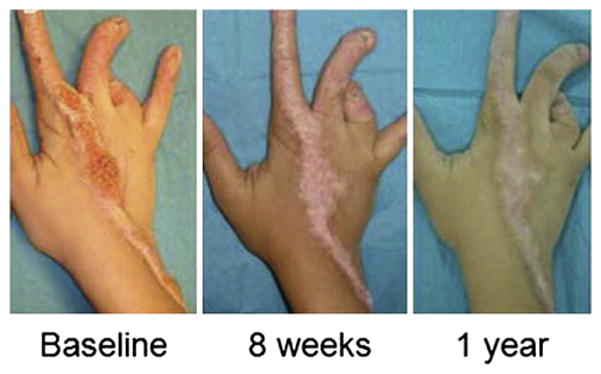
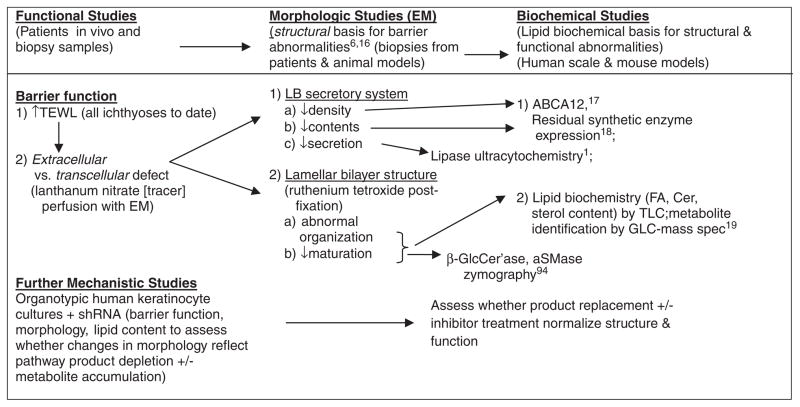
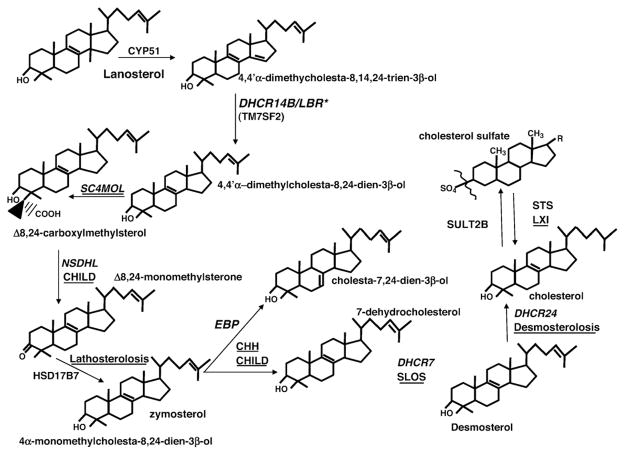
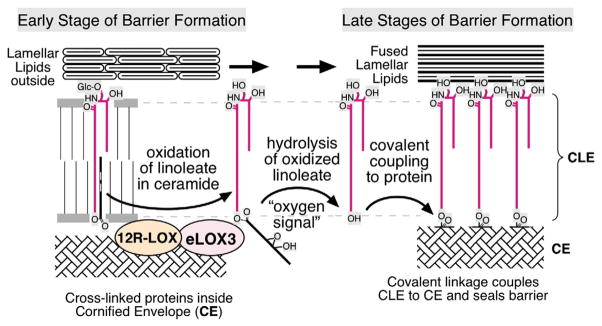
Ichthyoses, including inherited disorders of lipid metabolism, display a permeability barrier abnormality in which the severity of the clinical phenotype parallels the prominence of the barrier defect. The pathogenesis of the cutaneous phenotype represents the consequences of the mutation for epidermal function, coupled with a "best attempt" by affected epidermis to generate a competent barrier in a terrestrial environment. A compromised barrier in normal epidermis triggers a vigorous set of metabolic responses that rapidly normalizes function, but ichthyotic epidermis, which is inherently compromised, only partially succeeds in this effort. Unraveling mechanisms that account for barrier dysfunction in the ichthyoses has identified multiple, subcellular, and biochemical processes that contribute to the clinical phenotype. Current treatment of the ichthyoses remains largely symptomatic: directed toward reducing scale or corrective gene therapy. Reducing scale is often minimally effective. Gene therapy is impeded by multiple pitfalls, including difficulties in transcutaneous drug delivery, high costs, and discomfort of injections. We have begun to use information about disease pathogenesis to identify novel, pathogenesis-based therapeutic strategies for the ichthyoses. The clinical phenotype often reflects not only a deficiency of pathway end product due to reduced-function mutations in key synthetic enzymes but often also accumulation of proximal, potentially toxic metabolites. As a result, depending upon the identified pathomechanism(s) for each disorder, the accompanying ichthyosis can be treated by topical provision of pathway product (eg, cholesterol), with or without a proximal enzyme inhibitor (eg, simvastatin), to block metabolite production. Among the disorders of distal cholesterol metabolism, the cutaneous phenotype in Congenital Hemidysplasia with Ichthyosiform Erythroderma and Limb Defects (CHILD syndrome) and X-linked ichthyosis reflect metabolite accumulation and deficiency of pathway product (ie, cholesterol). We validated this therapeutic approach in two CHILD syndrome patients who failed to improve with topical cholesterol alone, but cleared with dual treatment with cholesterol plus lovastatin. In theory, the ichthyoses in other inherited lipid metabolic disorders could be treated analogously. This pathogenesis (pathway)-driven approach possesses several inherent advantages: (1) it is mechanism-specific for each disorder; (2) it is inherently safe, because natural lipids and/or approved drugs often are utilized; and (3) it should be inexpensive, and therefore it could be used widely in the developing world.
鱼鳞病,包括脂质代谢的遗传性疾病,表现出一种渗透性屏障异常,其临床表型的严重程度与屏障缺陷的明显程度平行。皮肤表型的发病机制代表了突变对表皮功能的影响,加上受影响的表皮在陆地环境中生成有能力的屏障的“最佳尝试”。正常表皮的屏障受损会引发一系列快速使功能正常化的代谢反应,但鱼鳞病表皮本身受损,只能在这方面部分成功。揭示鱼鳞病屏障功能障碍的机制已经确定了多个亚细胞和生化过程,这些过程有助于临床表型。目前鱼鳞病的治疗仍然主要是对症治疗:旨在减少鳞屑或进行矫正基因治疗。减少鳞屑的效果通常很小。基因治疗受到多种困难的阻碍,包括经皮药物输送困难、成本高和注射不适。我们已经开始利用疾病发病机制的信息来为鱼鳞病确定新的、基于发病机制的治疗策略。临床表型不仅反映了由于关键合成酶的功能丧失突变导致途径终产物的缺乏,而且还经常反映了潜在有毒代谢物的积累。因此,根据每种疾病的确定的病理机制,伴随的鱼鳞病可以通过局部提供途径产物(例如胆固醇)进行治疗,有或没有近端酶抑制剂(例如辛伐他汀)来阻止代谢产物的产生。在远端胆固醇代谢紊乱中,先天性半侧发育不良伴鱼鳞病样红皮病和肢体缺陷(CHILD 综合征)和 X 连锁鱼鳞病的皮肤表型反映了代谢物的积累和途径产物(即胆固醇)的缺乏。我们在两名未能单独用局部胆固醇改善的 CHILD 综合征患者中验证了这种治疗方法,但联合使用胆固醇加 lovastatin 治疗后得到了清除。理论上,其他遗传性脂质代谢紊乱的鱼鳞病可以类似地进行治疗。这种发病机制(途径)驱动的方法具有几个内在的优势:(1)它对每种疾病都是特定的机制;(2)它是内在安全的,因为天然脂质和/或批准的药物通常被利用;(3)它应该是廉价的,因此可以在发展中国家广泛使用。