Institute of Biomedical Sciences, Academia Sinica, Taipei, 11529, Taiwan, Republic of China.
J Biomed Sci. 2012 May 30;19(1):55. doi: 10.1186/1423-0127-19-55.
N-ethyl-N-nitrosourea mutagenesis was used to induce a point mutation in C57BL/6 J mice. Pain-related phenotype screening was performed in 915 G3 mice. We report the detection of a heritable recessive mutant in meiotic recombinant N1F1 mice that caused an abnormal pain sensitivity phenotype with spontaneous skin inflammation in the paws and ears.
We investigated abnormal sensory processing, neuronal peptides, and behavioral responses after the induction of autoinflammatory disease. Single-nucleotide polymorphism (SNP) markers and polymerase chain reaction product sequencing were used to identify the mutation site.
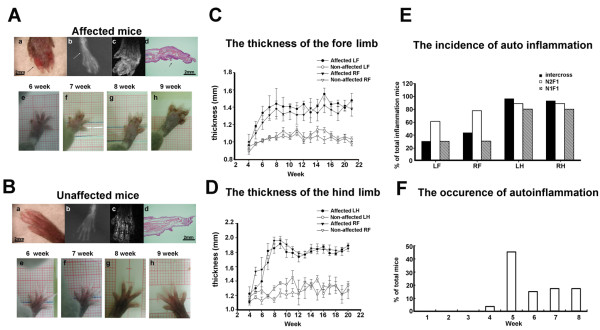
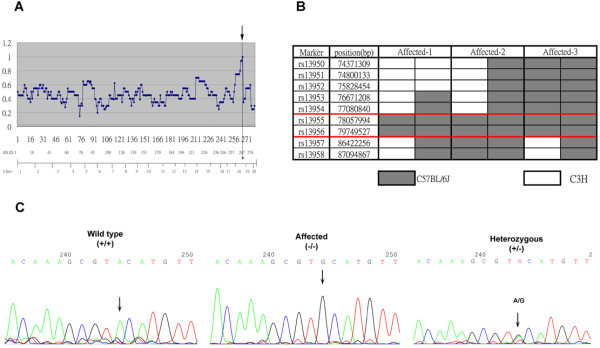
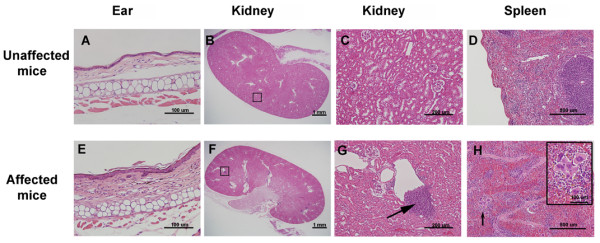
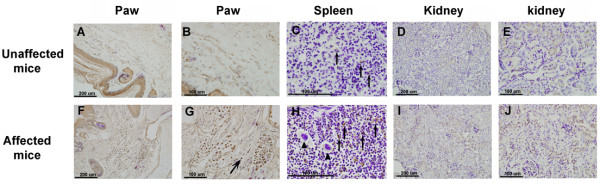
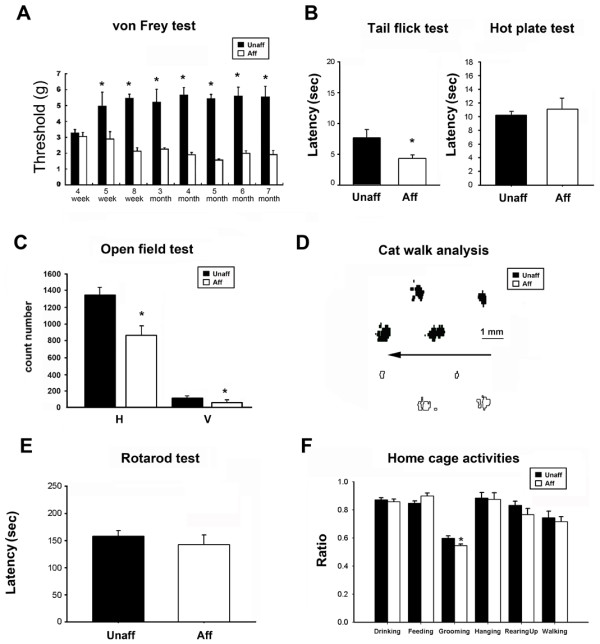
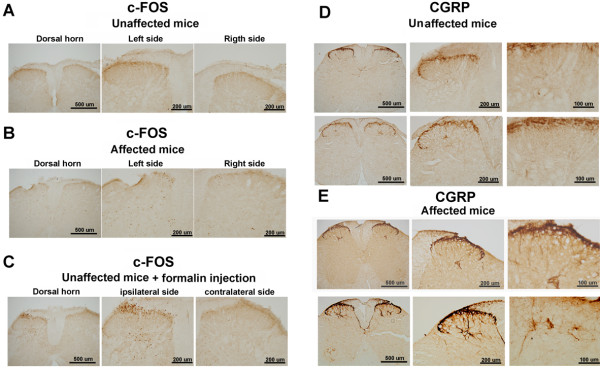
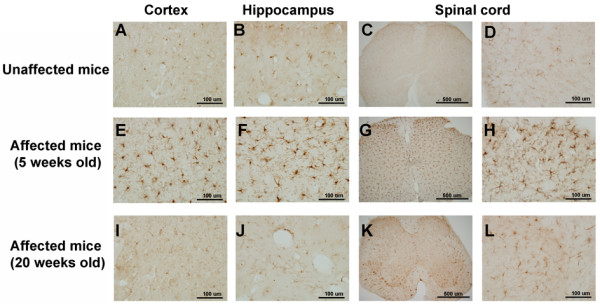
All affected mice developed paw inflammation at 4-8 weeks. Histological examinations revealed hyperplasia of the epidermis in the inflamed paws and increased macrophage expression in the spleen and paw tissues. Mechanical and thermal nociceptive response thresholds were reduced in the affected mice. Locomotor activity was decreased in affected mice with inflamed hindpaws, and this reduction was attributable to the avoidance of contact of the affected paw with the floor. Motor strength and daily activity in the home cage in the affected mice did not show any significant changes. Although Fos immunoreactivity was normal in the dorsal horn of affected mice, calcitonin gene-related peptide immunoreactivity significantly increased in the deep layer of the dorsal horn. The number of microglia increased in the spinal cord, hippocampus, and cerebral cortex in affected mice, and the proliferation of microglia was maintained for a couple of months. Two hundred eighty-five SNP markers were used to reveal the affected gene locus, which was found on the distal part of chromosome 18. A point mutation was detected at A to G in exon 8 of the pstpip2 gene, resulting in a conserved tyrosine residue at amino acid 180 replaced by cysteine (Y180 C).
The data provide definitive evidence that a mutation in pstpip2 causes autoinflammatory disease in an N-ethyl-N-nitrosourea mutagenesis mouse model. Thus, our pstpip2 mutant mice provide a new model for investigating the potential mechanisms of inflammatory pain.
利用 N-乙基-N-亚硝脲诱变剂在 C57BL/6J 小鼠中诱导点突变。在 915G3 小鼠中进行与疼痛相关的表型筛选。我们报告了在减数分裂重组 N1F1 小鼠中发现的一种可遗传隐性突变体,该突变体导致一种异常的疼痛敏感表型,伴有爪子和耳朵自发的皮肤炎症。
我们研究了自发炎症性疾病诱导后的异常感觉处理、神经元肽和行为反应。使用单核苷酸多态性 (SNP) 标记物和聚合酶链反应产物测序来确定突变位点。
所有受影响的小鼠在 4-8 周时出现爪子炎症。组织学检查显示,受影响爪子的表皮增生,脾脏和爪子组织中的巨噬细胞表达增加。受影响的小鼠机械和热伤害感受阈值降低。受影响的爪子有炎症的小鼠的运动活性降低,这种减少归因于避免受影响的爪子与地面接触。受影响的小鼠的运动力量和在笼内的日常活动没有显示出任何显著变化。虽然 Fos 免疫反应在受影响的小鼠背角正常,但降钙素基因相关肽免疫反应在背角的深层显著增加。受影响的小鼠脊髓、海马和大脑皮层中的小胶质细胞数量增加,小胶质细胞的增殖持续了几个月。使用 285 个 SNP 标记物揭示受影响的基因座,该基因座位于 18 号染色体的远端。在 pstpip2 基因的 8 号外显子中检测到 A 到 G 的点突变,导致氨基酸 180 处保守的酪氨酸残基被半胱氨酸取代(Y180C)。
数据提供了明确的证据,表明 pstpip2 的突变导致 N-乙基-N-亚硝脲诱变剂小鼠模型中的自炎症性疾病。因此,我们的 pstpip2 突变小鼠为研究炎症性疼痛的潜在机制提供了一个新的模型。