Department of Biomedical Imaging, Genentech Inc, 1 DNA Way, South San Francisco, CA, 94080, USA.
EJNMMI Res. 2012 May 31;2(1):22. doi: 10.1186/2191-219X-2-22.
The BRAF inhibitor, vemurafenib, has recently been approved for the treatment of metastatic melanoma in patients harboring BRAFV600 mutations. Currently, dual BRAF and MEK inhibition are ongoing in clinical trials with the goal of overcoming the acquired resistance that has unfortunately developed in some vemurafenib patients. FDG-PET measures of metabolic activity are increasingly employed as a pharmacodynamic biomarker for guiding single-agent or combination therapies by gauging initial drug response and monitoring disease progression. However, since tumors are inherently heterogeneous, investigating the effects of BRAF and MEK inhibition on FDG uptake in a panel of different melanomas could help interpret imaging outcomes.
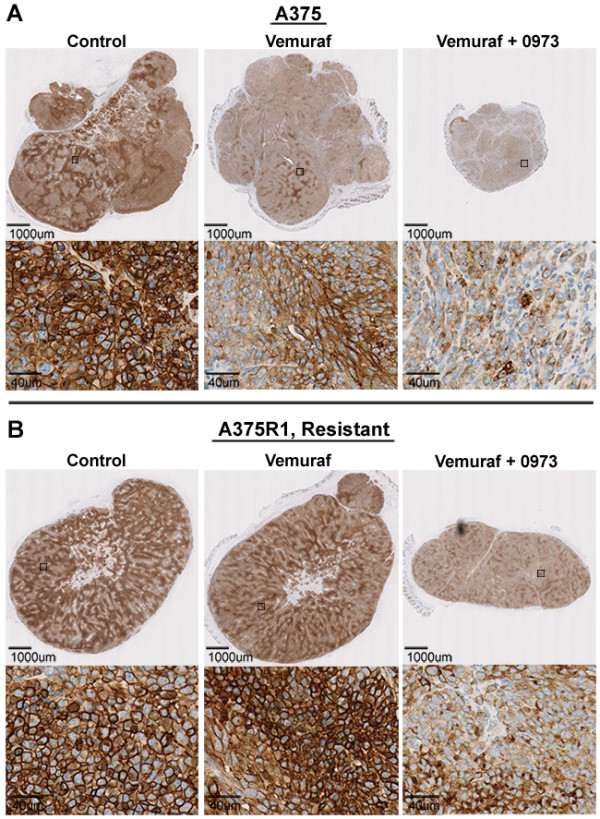
18 F-FDG uptake was measured in vitro in cells with wild-type and mutant (V600) BRAF, and in melanoma cells with an acquired resistance to vemurafenib. We treated the cells with vemurafenib alone or in combination with MEK inhibitor GDC-0973. PET imaging was used in mice to measure FDG uptake in A375 melanoma xenografts and in A375 R1, a vemurafenib-resistant derivative. Histological and biochemical studies of glucose transporters, the MAPK and glycolytic pathways were also undertaken.
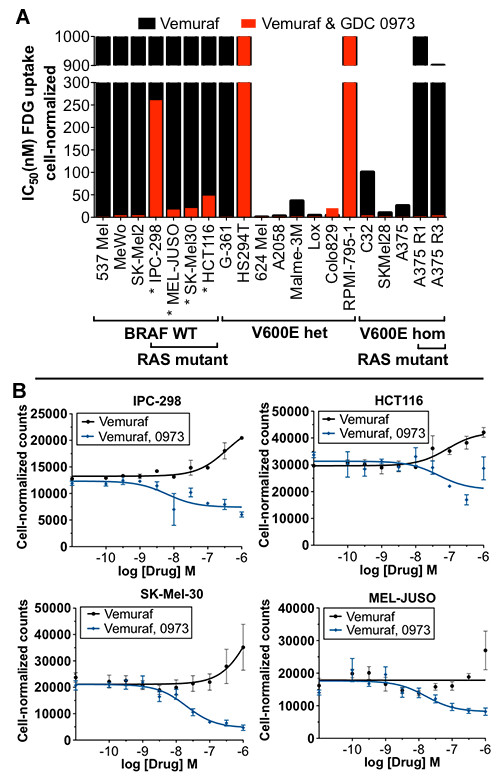
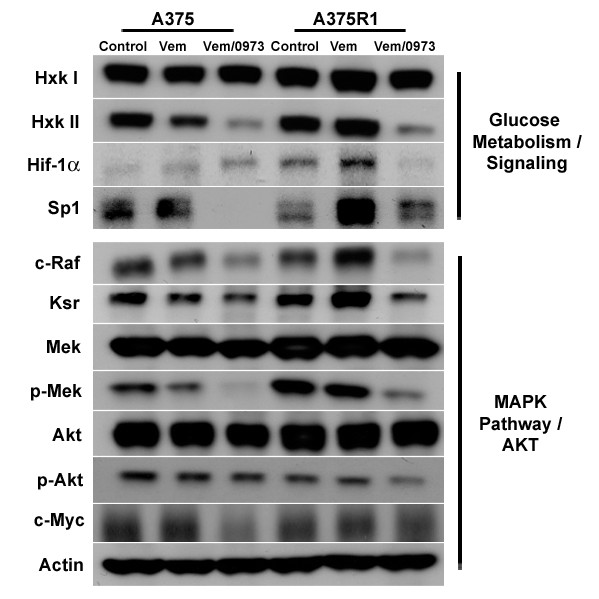
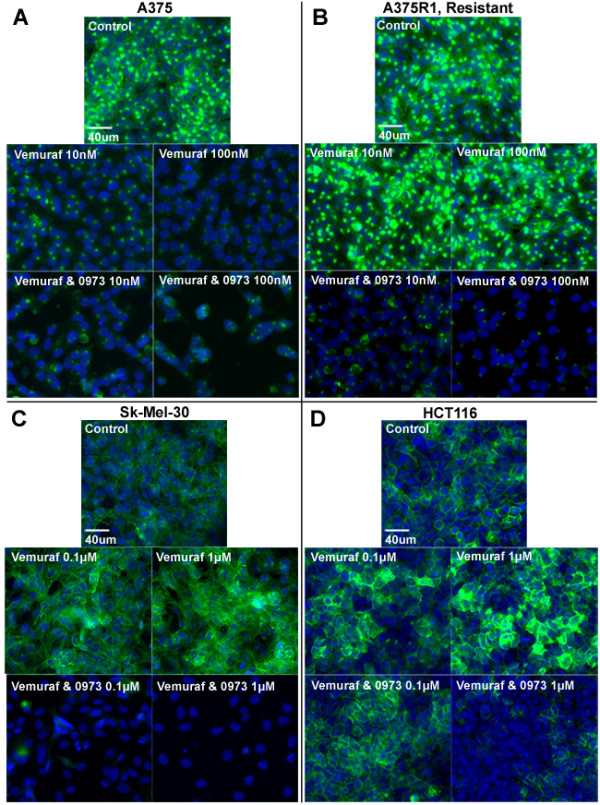
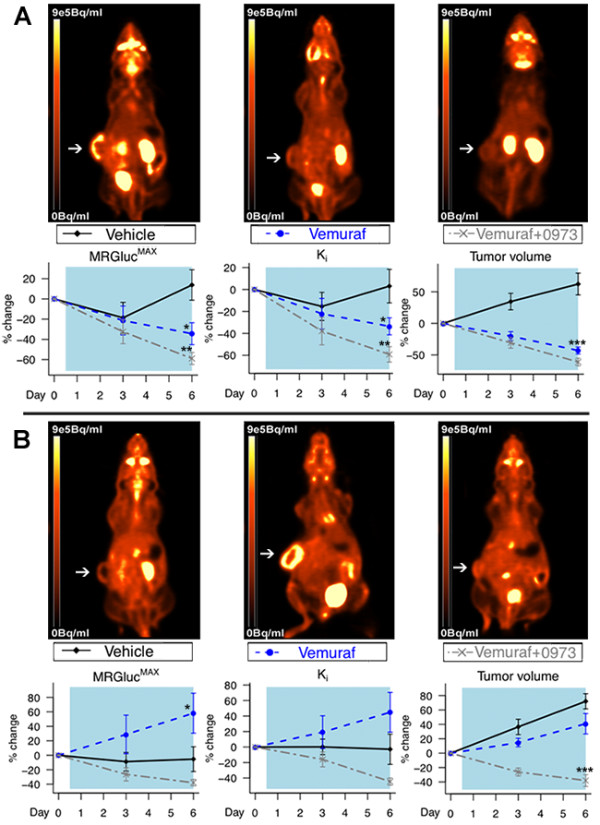
We demonstrate that vemurafenib is equally effective at reducing FDG uptake in cell lines harboring either heterozygous or homozygous BRAFV600 but ineffective in cells with acquired resistance or having WT BRAF status. However, combination with GDC-0973 results in a highly significant increase of efficacy and inhibition of FDG uptake across all twenty lines. Drug-induced changes in FDG uptake were associated with altered levels of membrane GLUT-1, and cell lines harboring RAS mutations displayed enhanced FDG uptake upon exposure to vemurafenib. Interestingly, we found that vemurafenib treatment in mice bearing drug-resistant A375 xenografts also induced increased FDG tumor uptake, accompanied by increases in Hif-1α, Sp1 and Ksr protein levels. Vemurafenib and GDC-0973 combination efficacy was associated with decreased levels of hexokinase II, c-RAF, Ksr and p-MEK protein.
We have demonstrated that 18 F-FDG-PET imaging reflects vemurafenib and GDC-0973 action across a wide range of metastatic melanomas. A delayed post-treatment increase in tumor FDG uptake should be considered carefully as it may well be an indication of acquired drug resistance.
ClinicalTrials.gov NCT01271803.
BRAF 抑制剂 vemurafenib 最近已被批准用于治疗携带 BRAFV600 突变的转移性黑色素瘤患者。目前,双重 BRAF 和 MEK 抑制正在临床试验中进行,目的是克服不幸在一些 vemurafenib 患者中出现的获得性耐药。FDG-PET 测量代谢活性越来越多地被用作指导单一药物或联合治疗的药效学生物标志物,通过衡量初始药物反应和监测疾病进展。然而,由于肿瘤本质上是异质的,因此研究 BRAF 和 MEK 抑制对一组不同黑色素瘤中 FDG 摄取的影响可以帮助解释成像结果。
在具有野生型和突变型(V600)BRAF 的细胞以及对 vemurafenib 产生获得性耐药的黑色素瘤细胞中,我们在体外测量 18F-FDG 摄取。我们单独用 vemurafenib 或联合 MEK 抑制剂 GDC-0973 治疗细胞。我们使用 PET 成像测量 A375 黑色素瘤异种移植瘤和 A375 R1(vemurafenib 耐药衍生物)中的 FDG 摄取。还进行了葡萄糖转运蛋白、MAPK 和糖酵解途径的组织学和生化研究。
我们证明,vemurafenib 对携带杂合或纯合 BRAFV600 的细胞系降低 FDG 摄取的效果相同,但对具有获得性耐药或具有 WT BRAF 状态的细胞无效。然而,联合使用 GDC-0973 可显著提高所有二十条曲线的疗效并抑制 FDG 摄取。药物诱导的 FDG 摄取变化与膜 GLUT-1 水平的改变有关,并且暴露于 vemurafenib 时携带 RAS 突变的细胞系摄取 FDG 的能力增强。有趣的是,我们发现,在携带耐药 A375 异种移植瘤的小鼠中,vemurafenib 治疗也诱导 FDG 肿瘤摄取增加,同时伴有 Hif-1α、Sp1 和 Ksr 蛋白水平升高。vemurafenib 和 GDC-0973 联合疗效与己糖激酶 II、c-RAF、Ksr 和 p-MEK 蛋白水平降低有关。
我们已经证明,18F-FDG-PET 成像反映了 vemurafenib 和 GDC-0973 在广泛的转移性黑色素瘤中的作用。治疗后肿瘤 FDG 摄取的延迟增加应仔细考虑,因为它很可能是获得性耐药的迹象。
ClinicalTrials.gov NCT01271803。