Department of Genetics, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil.
Evol Bioinform Online. 2012;8:207-18. doi: 10.4137/EBO.S9627. Epub 2012 May 14.
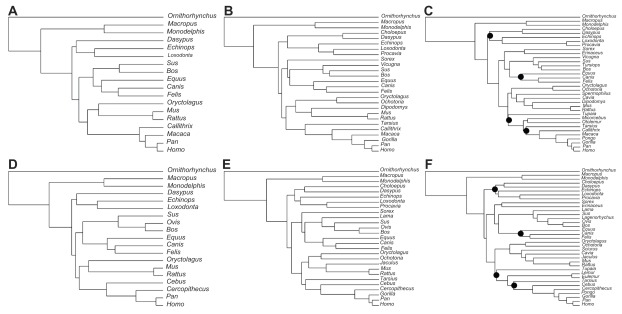
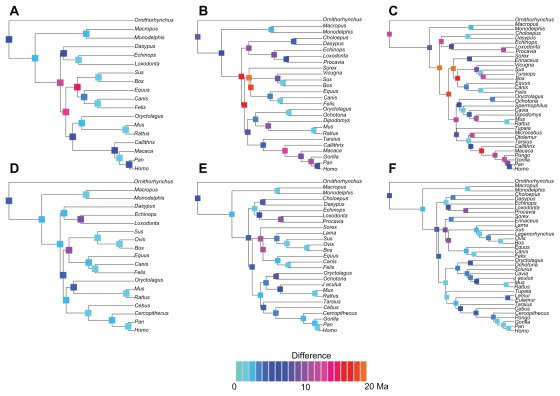
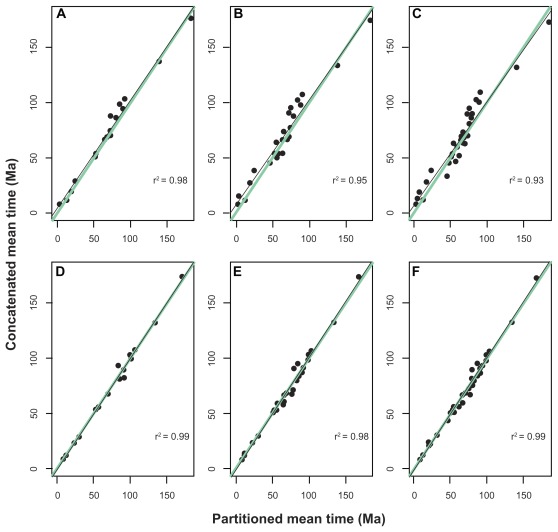
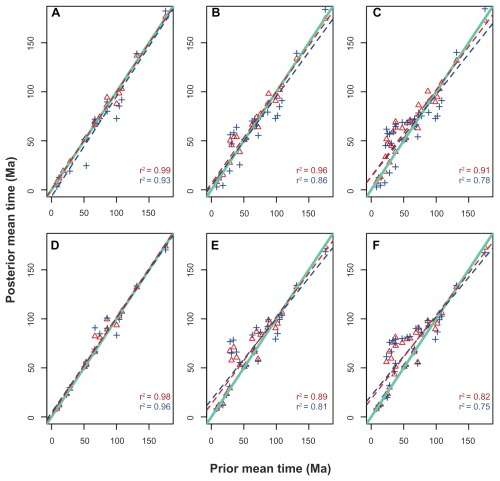
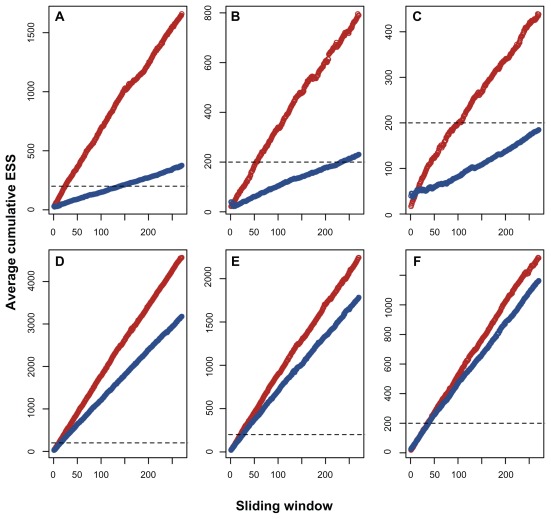
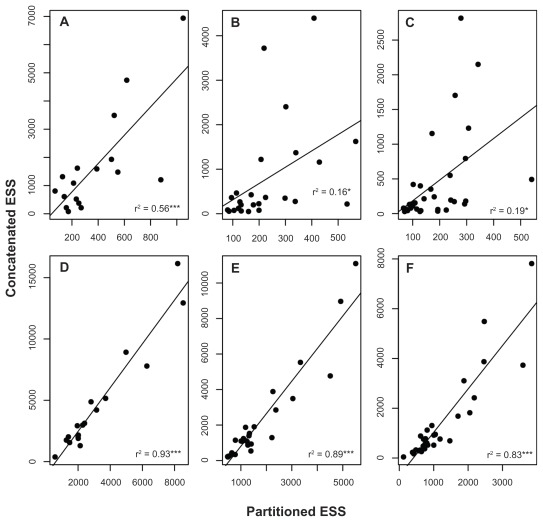
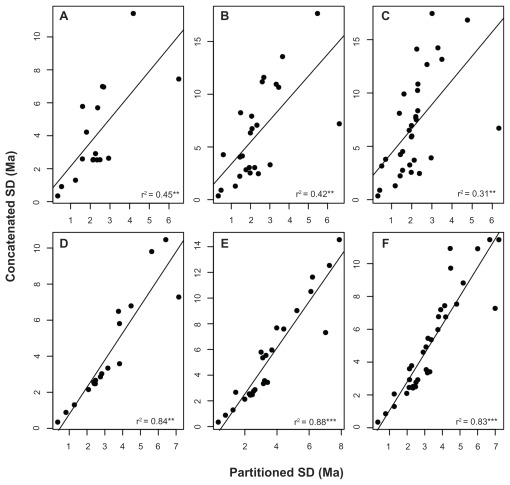
Data partitioning has long been regarded as an important parameter for phylogenetic inference. The division of heterogeneous multigene data sets into partitions with similar substitution patterns is known to increase the performance of probabilistic phylogenetic methods. However, the effect of the partitioning scheme on divergence time estimates has generally been ignored. To investigate the impact of data partitioning on the estimation of divergence times, we have constructed two genomic data sets. The first one with 15 nuclear genes comprising 50,928 bp were selected from the OrthoMam database; the second set was composed of complete mitochondrial genomes. We studied two partitioning schemes: concatenated supermatrices and partitioned gene analysis. We have also measured the impact of taxonomic sampling on the estimates. After drawing divergence time inferences using the uncorrelated relaxed clock in BEAST, we have compared the age estimates between the partitioning schemes. Our results show that, in general, both schemes resulted in similar chronological estimates, however the concatenated data sets were more efficient than the partitioned ones in attaining suitable effective sample sizes.
数据分区长期以来一直被视为系统发育推断的一个重要参数。将异质多基因数据集划分为具有相似替代模式的分区,已知可以提高概率系统发育方法的性能。然而,分区方案对分歧时间估计的影响通常被忽略。为了研究数据分区对分歧时间估计的影响,我们构建了两个基因组数据集。第一个数据集由 OrthoMam 数据库中包含 50928bp 的 15 个核基因组成;第二个数据集由完整的线粒体基因组组成。我们研究了两种分区方案:串联超矩阵和分区基因分析。我们还测量了分类采样对估计的影响。在使用 BEAST 中的无相关松弛时钟进行分歧时间推断后,我们比较了分区方案之间的年龄估计值。我们的结果表明,一般来说,两种方案都产生了相似的年代估计值,然而,串联数据集在获得合适的有效样本量方面比分区数据集更有效。