Departamento de Ciencia y Tecnología, Universidad Nacional de Quilmes, Buenos Aires, Argentina.
BMC Genomics. 2012 Jun 18;13 Suppl 4(Suppl 4):S5. doi: 10.1186/1471-2164-13-S4-S5.
Non-synonymous coding SNPs (nsSNPs) that are associated to disease can also be related with alterations in protein stability. Computational methods are available to predict the effect of single amino acid substitutions (SASs) on protein stability based on a single folded structure. However, the native state of a protein is not unique and it is better represented by the ensemble of its conformers in dynamic equilibrium. The maintenance of the ensemble is essential for protein function. In this work we investigated how protein conformational diversity can affect the discrimination of neutral and disease related SASs based on protein stability estimations. For this purpose, we used 119 proteins with 803 associated SASs, 60% of which are disease related. Each protein was associated with its corresponding set of available conformers as found in the Protein Conformational Database (PCDB). Our dataset contains proteins with different extensions of conformational diversity summing up a total number of 1023 conformers.
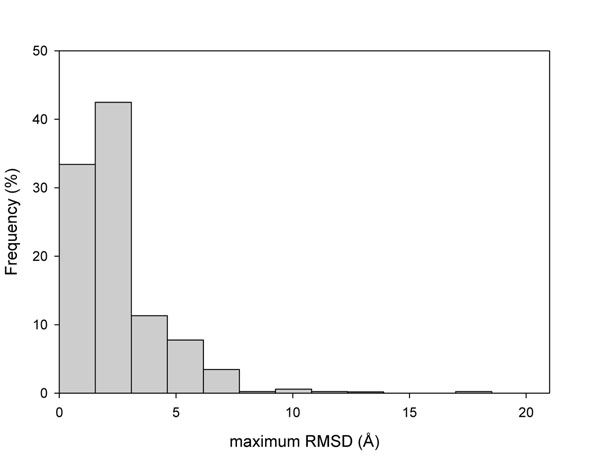
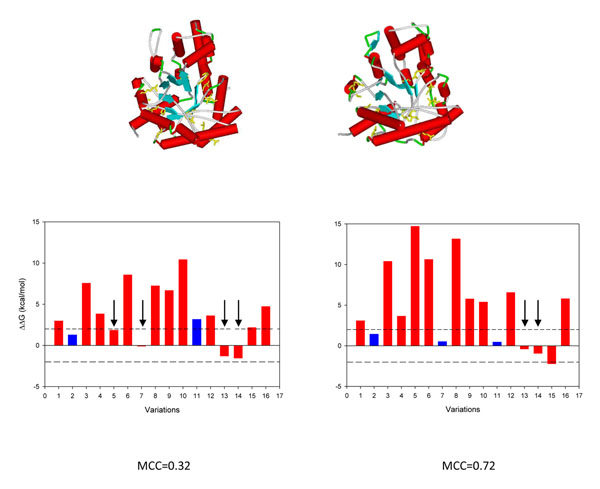
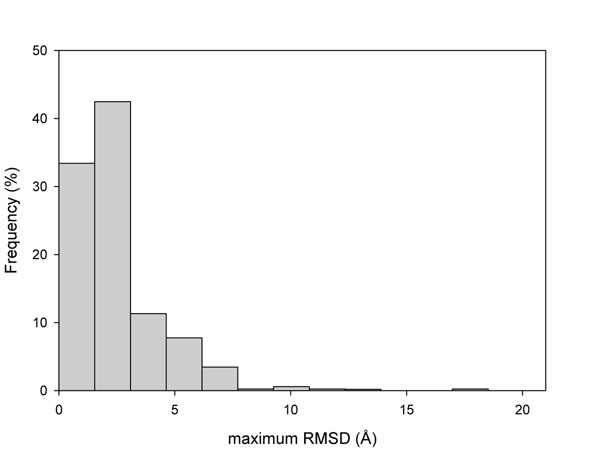
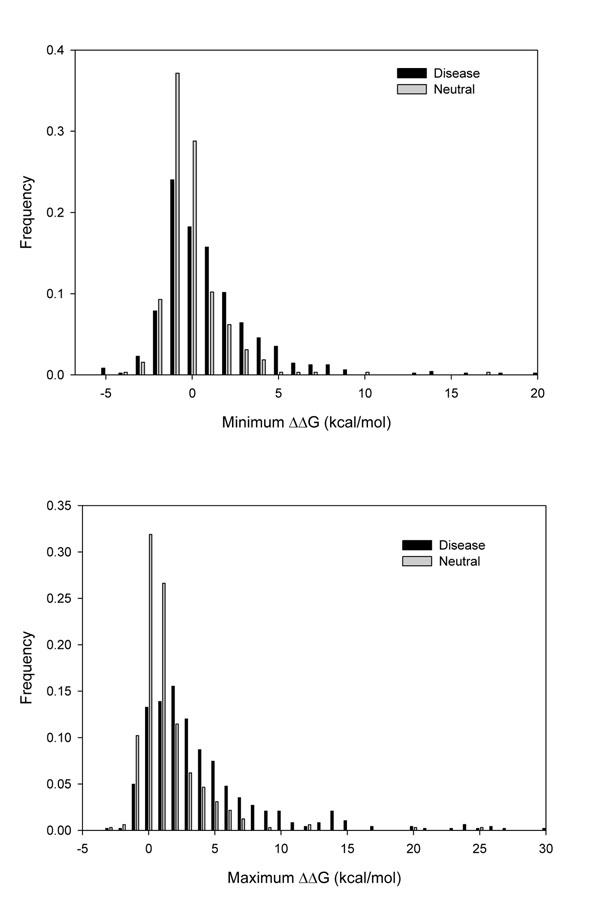
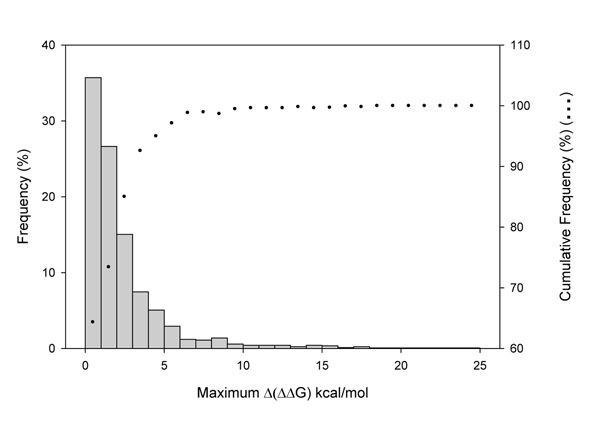
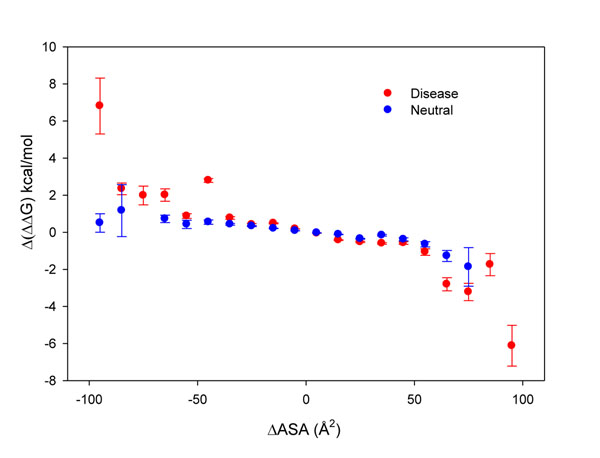
The existence of different conformers for a given protein introduces great variability in the estimation of the protein stability (ΔΔG) after a single amino acid substitution (SAS) as computed with FoldX. Indeed, in 35% of our protein set at least one SAS can be described as stabilizing, destabilizing or neutral when a cutoff value of ±2 kcal/mol is adopted for discriminating neutral from perturbing SASs. However, when the ΔΔG variability among conformers is taken into account, the correlation among the perturbation of protein stability and the corresponding disease or neutral phenotype increases as compared with the same analysis on single protein structures. At the conformer level, we also found that the different conformers correlate in a different way to the corresponding phenotype.
Our results suggest that the consideration of conformational diversity can improve the discrimination of neutral and disease related protein SASs based on the evaluation of the corresponding Gibbs free energy change.
与疾病相关的非同义编码单核苷酸多态性(nsSNP)也可能与蛋白质稳定性的改变有关。现已有一些计算方法可基于单一折叠结构预测单一氨基酸取代(SAS)对蛋白质稳定性的影响。然而,蛋白质的天然状态并不是唯一的,它更好地由其在动态平衡中的构象异构体的集合来表示。构象异构体集合的维持对于蛋白质的功能至关重要。在这项工作中,我们研究了蛋白质构象多样性如何影响基于蛋白质稳定性估计的中性和疾病相关 SAS 的区分。为此,我们使用了 119 种蛋白质,这些蛋白质与 803 个相关的 SAS 相关,其中 60%与疾病相关。每个蛋白质都与在蛋白质构象数据库(PCDB)中发现的其相应的可用构象集合相关联。我们的数据集包含具有不同构象多样性扩展的蛋白质,总计有 1023 个构象。
对于给定的蛋白质,不同构象的存在导致在使用 FoldX 计算单一氨基酸取代(SAS)后对蛋白质稳定性(ΔΔG)的估计存在很大的变异性。实际上,在我们的蛋白质组的 35%中,当采用±2 kcal/mol 的截断值来区分中性和干扰 SAS 时,至少有一个 SAS 可以被描述为稳定、不稳定或中性。然而,当考虑构象异构体之间的 ΔΔG 变异性时,与稳定性扰动相关的蛋白质与相应的疾病或中性表型之间的相关性会增加,与对单一蛋白质结构的相同分析相比。在构象水平上,我们还发现不同的构象以不同的方式与相应的表型相关。
我们的结果表明,考虑构象多样性可以提高基于评估相应吉布斯自由能变化的中性和疾病相关蛋白质 SAS 的区分。