Department of Molecular Cell Biology, Center for Systems Biology, University of Texas at Dallas, Richardson, Texas, United States of America.
PLoS One. 2012;7(6):e38112. doi: 10.1371/journal.pone.0038112. Epub 2012 Jun 29.
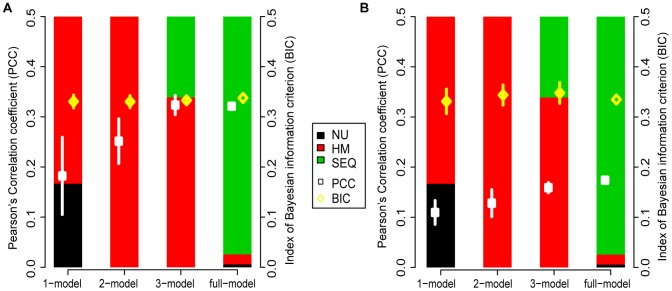
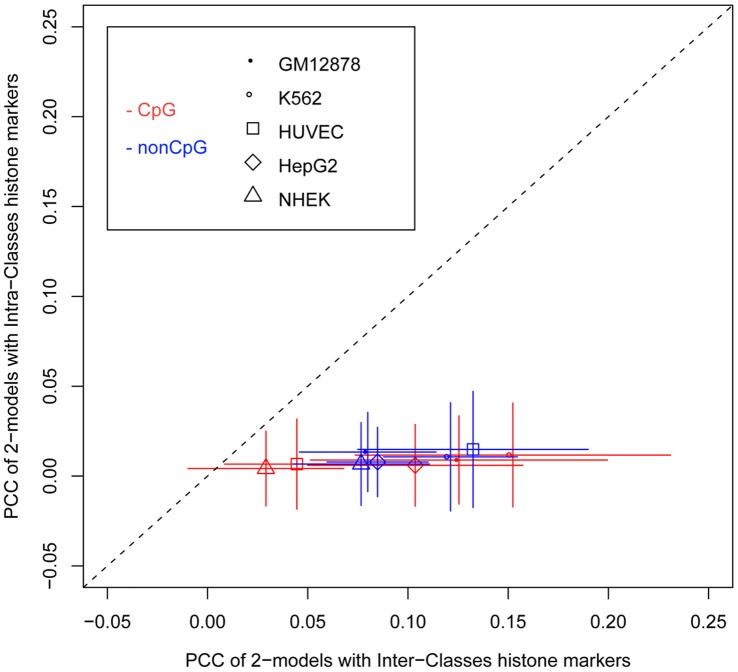
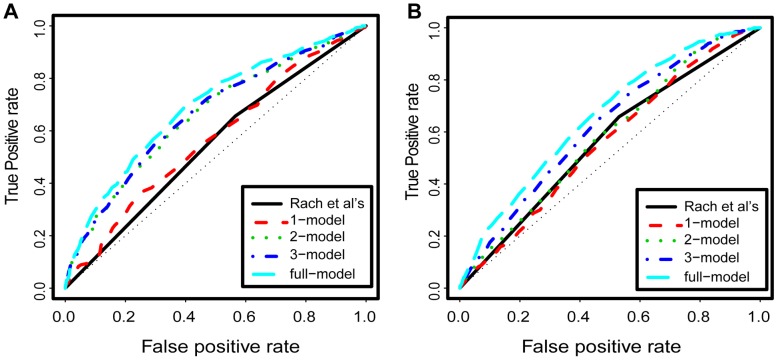
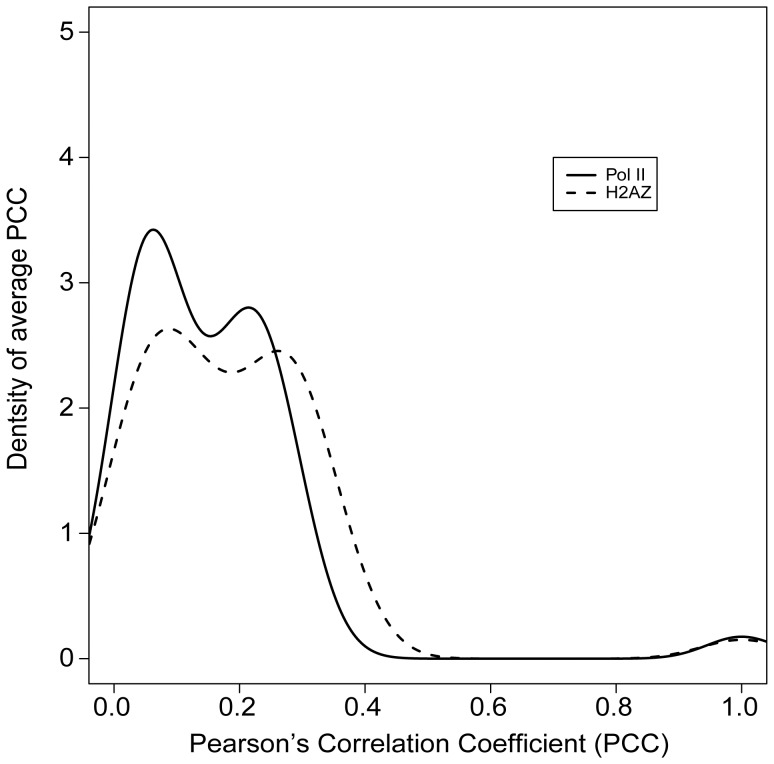
Deep sequencing of 5' capped transcripts has revealed a variety of transcription initiation patterns, from narrow, focused promoters to wide, broad promoters. Attempts have already been made to model empirically classified patterns, but virtually no quantitative models for transcription initiation have been reported. Even though both genetic and epigenetic elements have been associated with such patterns, the organization of regulatory elements is largely unknown. Here, linear regression models were derived from a pool of regulatory elements, including genomic DNA features, nucleosome organization, and histone modifications, to predict the distribution of transcription start sites (TSS). Importantly, models including both active and repressive histone modification markers, e.g. H3K4me3 and H4K20me1, were consistently found to be much more predictive than models with only single-type histone modification markers, indicating the possibility of "bivalent-like" epigenetic control of transcription initiation. The nucleosome positions are proposed to be coded in the active component of such bivalent-like histone modification markers. Finally, we demonstrated that models trained on one cell type could successfully predict TSS distribution in other cell types, suggesting that these models may have a broader application range.
对 5' 帽状转录物的深度测序揭示了各种转录起始模式,从狭窄、集中的启动子到广泛、广泛的启动子。已经有人试图对经验分类的模式进行建模,但实际上还没有报道用于转录起始的定量模型。尽管遗传和表观遗传因素都与这些模式有关,但调控元件的组织在很大程度上是未知的。在这里,线性回归模型是从包括基因组 DNA 特征、核小体组织和组蛋白修饰在内的一组调控元件中得出的,以预测转录起始位点 (TSS) 的分布。重要的是,包括活性和抑制性组蛋白修饰标记物(例如 H3K4me3 和 H4K20me1)的模型被发现比仅具有单一类型组蛋白修饰标记物的模型更具预测性,这表明转录起始的“双价样”表观遗传控制的可能性。提出核小体位置编码在这种双价样组蛋白修饰标记物的活性成分中。最后,我们证明了在一种细胞类型上训练的模型可以成功预测其他细胞类型中的 TSS 分布,这表明这些模型可能具有更广泛的应用范围。