Department of Biochemistry and Microbiology, Faculty of Food and Biochemical Technology, Institute of Chemical Technology Prague, Prague, Czech Republic.
PLoS One. 2012;7(7):e40653. doi: 10.1371/journal.pone.0040653. Epub 2012 Jul 13.
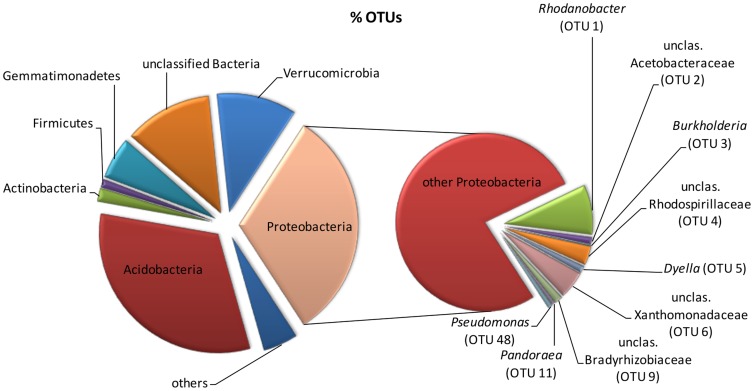
Bacteria were identified associated with biodegradation of aromatic pollutants biphenyl, benzoate, and naphthalene in a long-term polychlorinated biphenyl- and polyaromatic hydrocarbon-contaminated soil. In order to avoid biases of culture-based approaches, stable isotope probing was applied in combination with sequence analysis of 16 S rRNA gene pyrotags amplified from (13)C-enriched DNA fractions. Special attention was paid to pyrosequencing data analysis in order to eliminate the errors caused by either generation of amplicons (random errors caused by DNA polymerase, formation of chimeric sequences) or sequencing itself. Therefore, sample DNA was amplified, sequenced, and analyzed along with the DNA of a mock community constructed out of 8 bacterial strains. This warranted that appropriate tools and parameters were chosen for sequence data processing. (13)C-labeled metagenomes isolated after the incubation of soil samples with all three studied aromatics were largely dominated by Proteobacteria, namely sequences clustering with the genera Rhodanobacter Burkholderia, Pandoraea, Dyella as well as some Rudaea- and Skermanella-related ones. Pseudomonads were mostly labeled by (13)C from naphthalene and benzoate. The results of this study show that many biphenyl/benzoate-assimilating bacteria derive carbon also from naphthalene, pointing out broader biodegradation abilities of some soil microbiota. The results also demonstrate that, in addition to traditionally isolated genera of degradative bacteria, yet-to-be cultured bacteria are important players in bioremediation. Overall, the study contributes to our understanding of biodegradation processes in contaminated soil. At the same time our results show the importance of sequencing and analyzing a mock community in order to more correctly process and analyze sequence data.
在长期受多氯联苯和多环芳烃污染的土壤中,鉴定出了与联苯、苯甲酸和萘生物降解有关的细菌。为了避免基于培养的方法的偏见,采用稳定同位素探测法结合 16S rRNA 基因焦磷酸测序从(13)C 富集 DNA 片段进行序列分析。特别关注焦磷酸测序数据分析,以消除由扩增子产生的误差(由 DNA 聚合酶引起的随机误差,形成嵌合序列)或测序本身引起的误差。因此,沿 8 株细菌构建的模拟群落的 DNA 进行扩增、测序和分析。这保证了为序列数据处理选择了适当的工具和参数。在用三种研究的芳烃孵育土壤样品后分离的(13)C 标记宏基因组主要由变形菌门(Proteobacteria)主导,即与 Rhodanobacter、Burkholderia、Pandoraea、Dyella 属以及一些与 Rudaea 和 Skermanella 相关的序列聚类。假单胞菌主要由萘和苯甲酸的(13)C 标记。该研究结果表明,许多联苯/苯甲酸同化细菌也从萘中获取碳,这表明一些土壤微生物群落具有更广泛的生物降解能力。研究结果还表明,除了传统上分离的降解细菌属外,尚未培养的细菌也是生物修复的重要参与者。总的来说,该研究有助于我们了解污染土壤中的生物降解过程。同时,我们的结果表明,测序和分析模拟群落对于更正确地处理和分析序列数据非常重要。