Pflug Alexander, Johnson Kenneth A, Engh Richard A
Norwegian Structural Biology Centre, Department of Chemistry, University of Tromsø, N-9037 Tromsø, Norway.
Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012 Aug 1;68(Pt 8):873-7. doi: 10.1107/S1744309112028655. Epub 2012 Jul 26.
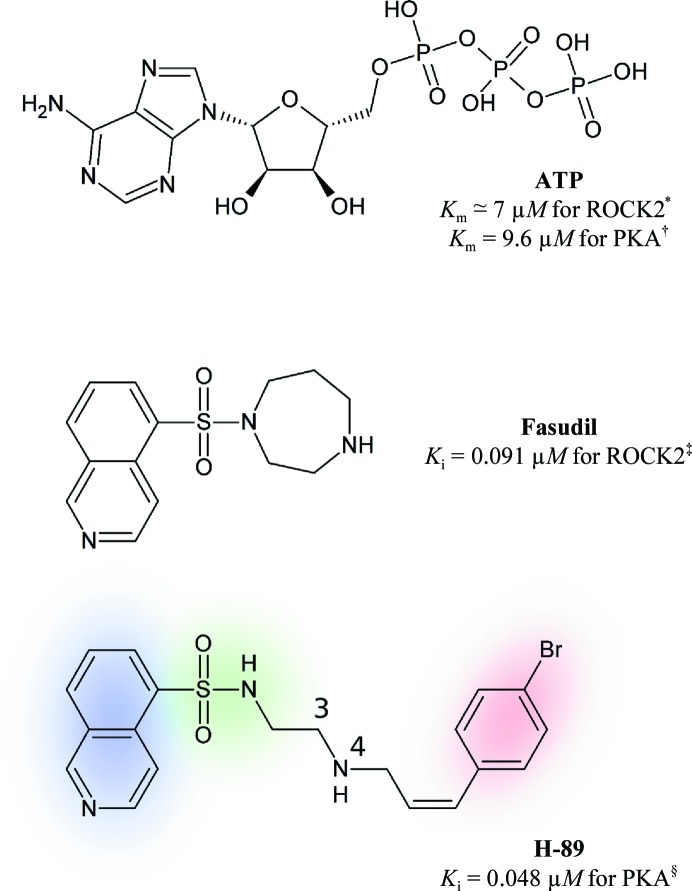
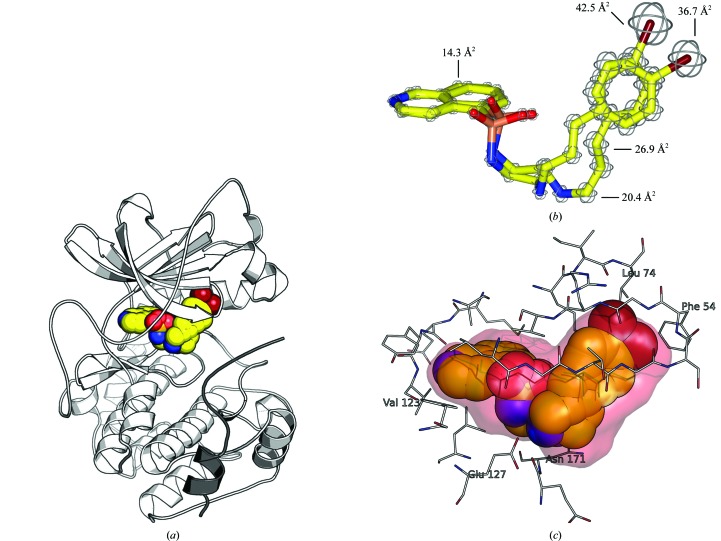
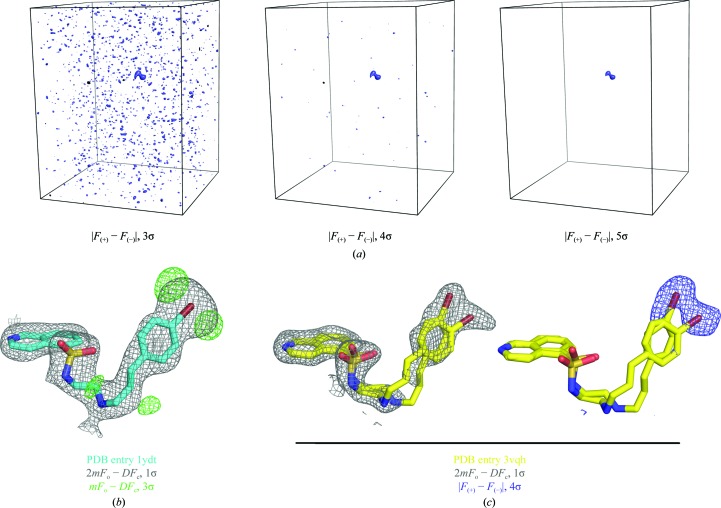
With its ability to show the interactions between drug-target proteins and small-molecule ligands, X-ray crystallography is an essential tool in drug-discovery programmes. However, its usefulness can be limited by crystallization artifacts or by the data resolution, and in particular when assumptions of unimodal binding (and isotropic motion) do not apply. Discrepancies between the modelled crystal structure and the physiological range of structures generally prevent quantitative estimation of binding energies. Improved crystal structure resolution will often not aid energy estimation because the conditions which provide the highest rigidity and resolution are not likely to reflect physiological conditions. Instead, strategies must be employed to measure and model flexibility and multiple binding modes to supplement crystallographic information. One useful tool is the use of anomalous dispersion for small molecules that contain suitable atoms. Here, an analysis of the binding of the kinase inhibitor H-89 to protein kinase A (PKA) is presented. H-89 contains a bromobenzene moiety that apparently binds with multiple conformations in the kinase ATP pocket. Using anomalous dispersion methods, it was possible to resolve these conformations into two distinct binding geometries.
由于能够展示药物靶点蛋白与小分子配体之间的相互作用,X射线晶体学是药物研发项目中的一项重要工具。然而,其效用可能会受到结晶假象或数据分辨率的限制,尤其是在单峰结合(以及各向同性运动)假设不适用的情况下。建模晶体结构与结构生理范围之间的差异通常会妨碍对结合能进行定量估计。提高晶体结构分辨率往往无助于能量估计,因为提供最高刚性和分辨率的条件不太可能反映生理条件。相反,必须采用策略来测量和模拟灵活性及多种结合模式,以补充晶体学信息。一个有用的工具是对含有合适原子的小分子使用反常色散。在此,展示了对激酶抑制剂H - 89与蛋白激酶A(PKA)结合的分析。H - 89含有一个溴苯基团,该基团显然在激酶ATP口袋中以多种构象结合。使用反常色散方法,可以将这些构象解析为两种不同的结合几何结构。