Oelmeier Stefan A, Dismer Florian, Hubbuch Jürgen
Karlsruhe Institute of Technology (KIT), Institute of Process Engineering in Life Sciences, Section IV: Biomolecular Separation Engineering, Karlsruhe, Germany.
BMC Biophys. 2012 Aug 8;5:14. doi: 10.1186/2046-1682-5-14.
Molecular Dynamics (MD) simulations are a promising tool to generate molecular understanding of processes related to the purification of proteins. Polyethylene glycols (PEG) of various length are commonly used in the production and purification of proteins. The molecular mechanisms behind PEG driven precipitation, aqueous two-phase formation or the effects of PEGylation are however still poorly understood.
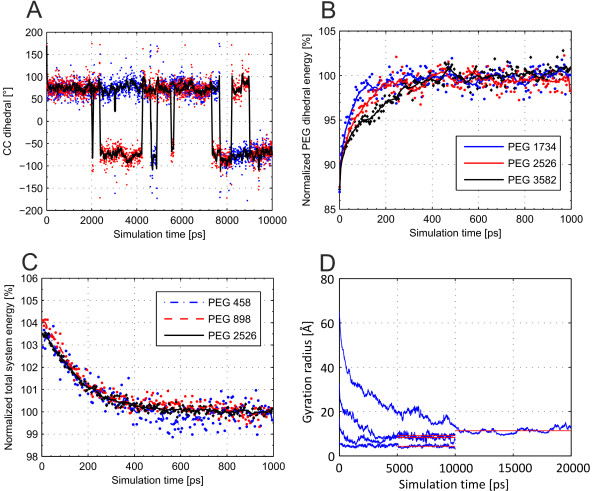
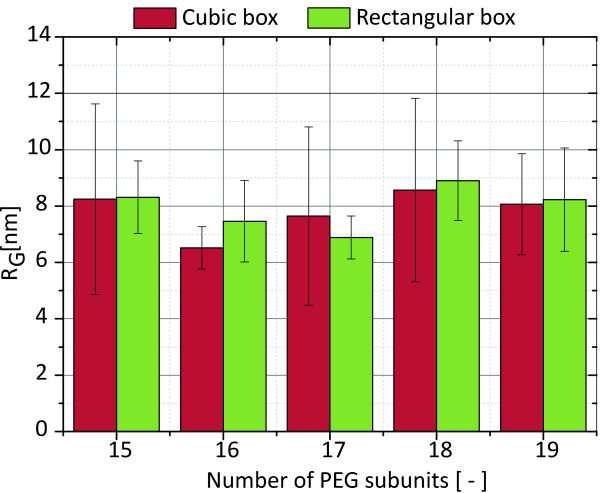
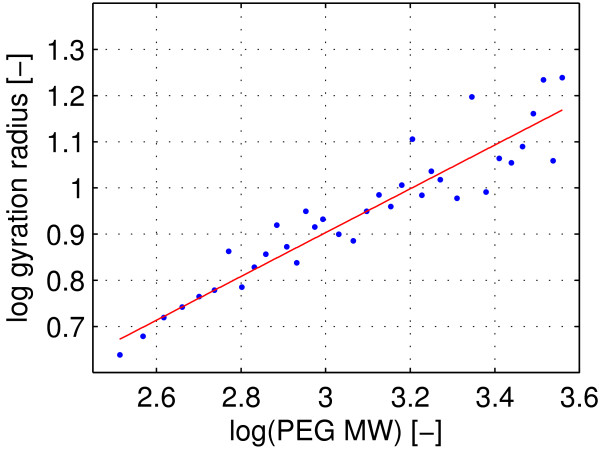
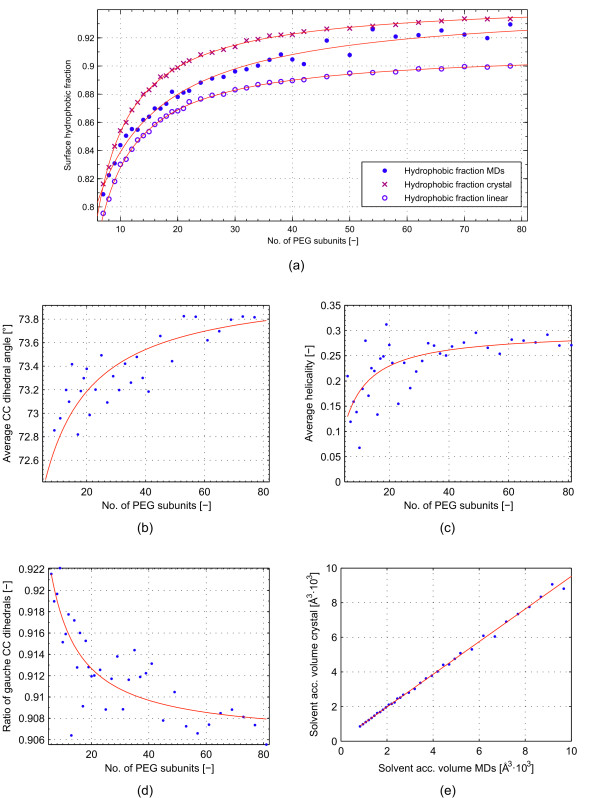
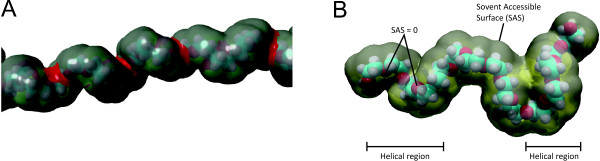
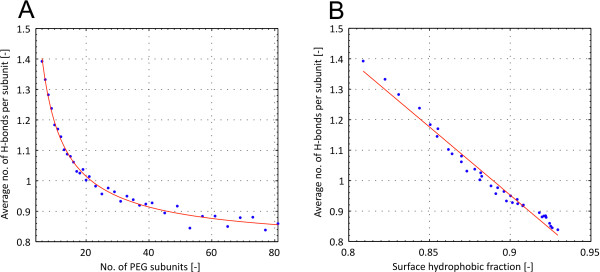
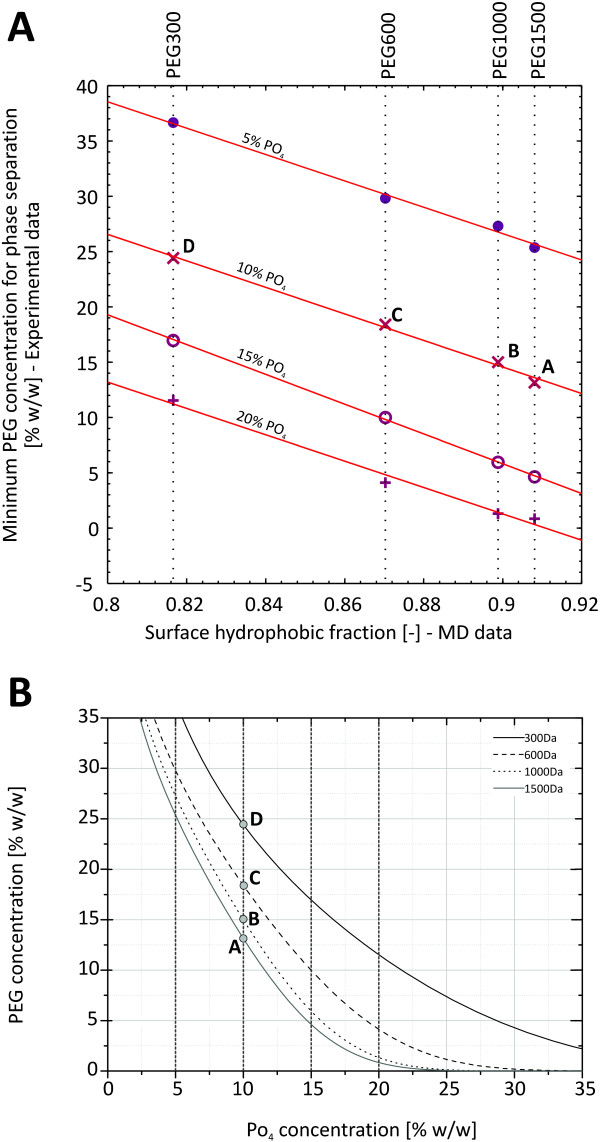
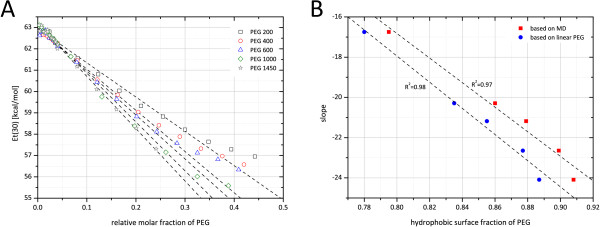
In this paper, we ran MD simulations of single PEG molecules of variable length in explicitly simulated water. The resulting structures are in good agreement with experimentally determined 3D structures of PEG. The increase in surface hydrophobicity of PEG of longer chain length could be explained on an atomic scale. PEG-water interactions as well as aqueous two-phase formation in the presence of PO4 were found to be correlated to PEG surface hydrophobicity.
We were able to show that the taken MD simulation approach is capable of generating both structural data as well as molecule descriptors in agreement with experimental data. Thus, we are confident of having a good in silico representation of PEG.
分子动力学(MD)模拟是一种很有前景的工具,可用于从分子层面理解与蛋白质纯化相关的过程。不同长度的聚乙二醇(PEG)常用于蛋白质的生产和纯化。然而,PEG驱动沉淀、水相两相形成或聚乙二醇化作用背后的分子机制仍知之甚少。
在本文中,我们对不同长度的单个PEG分子在显式模拟水中进行了MD模拟。所得结构与实验测定的PEG三维结构高度吻合。长链PEG表面疏水性的增加可以在原子尺度上得到解释。发现PEG与水的相互作用以及在存在PO4的情况下水相两相的形成与PEG表面疏水性相关。
我们能够证明所采用的MD模拟方法能够生成与实验数据一致的结构数据和分子描述符。因此,我们相信对PEG有良好的计算机模拟表示。