Institute Born-Bunge, University of Antwerp, Antwerpen, Belgium.
Acta Neuropathol. 2012 Sep;124(3):353-72. doi: 10.1007/s00401-012-1029-x. Epub 2012 Aug 14.
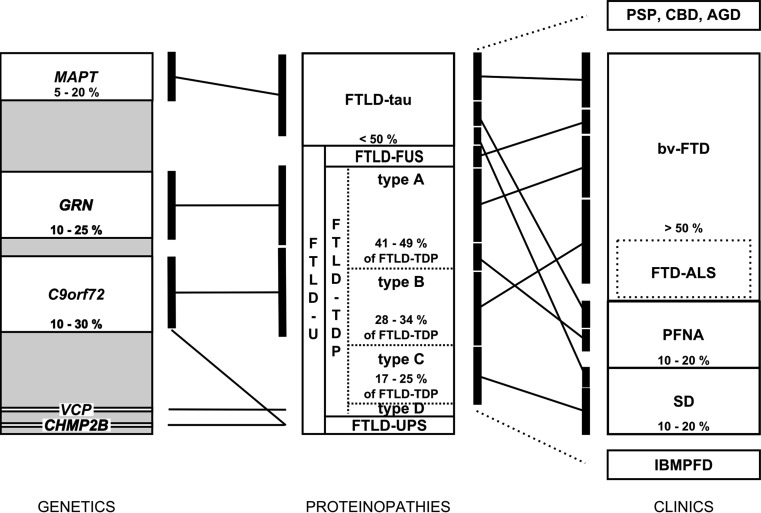
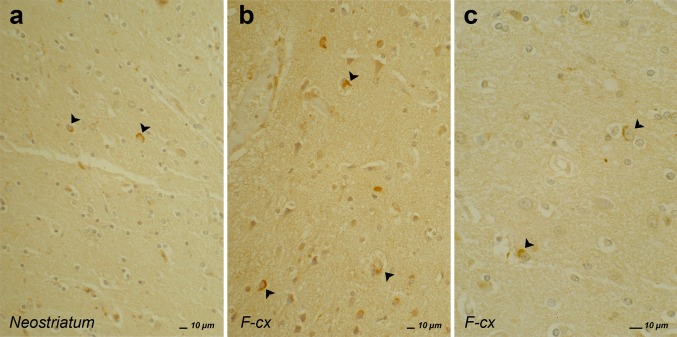
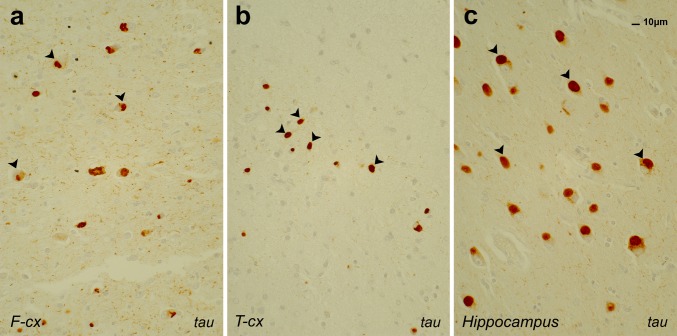
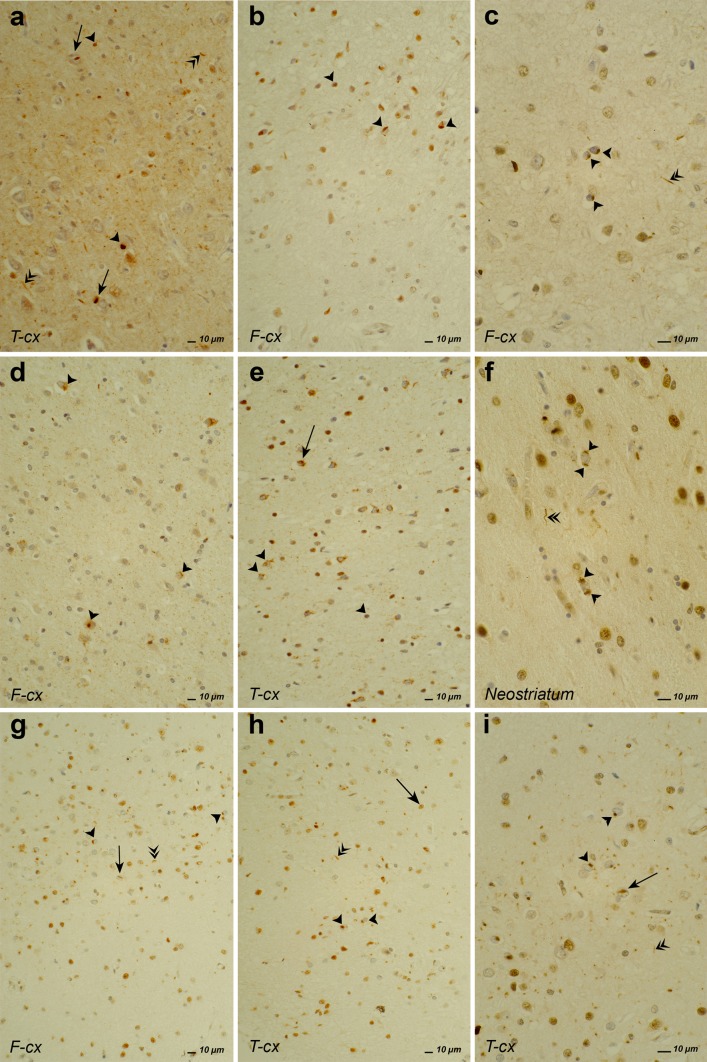
Frontotemporal lobar degeneration (FTLD) is a heterogeneous group of disorders characterized by disturbances of behavior and personality and different types of language impairment with or without concomitant features of motor neuron disease or parkinsonism. FTLD is characterized by atrophy of the frontal and anterior temporal brain lobes. Detailed neuropathological studies have elicited proteinopathies defined by inclusions of hyperphosphorylated microtubule-associated protein tau, TAR DNA-binding protein TDP-43, fused-in-sarcoma or yet unidentified proteins in affected brain regions. Rather than the type of proteinopathy, the site of neurodegeneration correlates relatively well with the clinical presentation of FTLD. Molecular genetic studies identified five disease genes, of which the gene encoding the tau protein (MAPT), the growth factor precursor gene granulin (GRN), and C9orf72 with unknown function are most frequently mutated. Rare mutations were also identified in the genes encoding valosin-containing protein (VCP) and charged multivesicular body protein 2B (CHMP2B). These genes are good markers to distinguish underlying neuropathological phenotypes. Due to the complex landscape of FTLD diseases, combined characterization of clinical, imaging, biological and genetic biomarkers is essential to establish a detailed diagnosis. Although major progress has been made in FTLD research in recent years, further studies are needed to completely map out and correlate the clinical, pathological and genetic entities, and to understand the underlying disease mechanisms. In this review, we summarize the current state of the rapidly progressing field of genetic, neuropathological and clinical research of this intriguing condition.
额颞叶变性(FTLD)是一组异质性疾病,其特征为行为和人格障碍以及不同类型的语言障碍,伴有或不伴有运动神经元病或帕金森病的特征。FTLD 的特征是额和前颞叶脑区萎缩。详细的神经病理学研究已经确定了蛋白病,其特征为在受影响的脑区中存在过度磷酸化的微管相关蛋白 tau、TAR DNA 结合蛋白 TDP-43、融合肉瘤或尚未鉴定的蛋白的包涵体。与蛋白病的类型相比,神经退行性变的部位与 FTLD 的临床表现相对较好地相关。分子遗传学研究鉴定了五个疾病基因,其中编码 tau 蛋白(MAPT)的基因、生长因子前体基因颗粒蛋白(GRN)和具有未知功能的 C9orf72 基因最常发生突变。还在编码包含缬氨酸的蛋白(VCP)和带电荷的多泡体蛋白 2B(CHMP2B)的基因中鉴定出罕见突变。这些基因是区分潜在神经病理学表型的良好标志物。由于 FTLD 疾病的复杂景观,临床、影像学、生物学和遗传生物标志物的综合特征对于建立详细诊断至关重要。尽管近年来 FTLD 研究取得了重大进展,但仍需要进一步研究以完全描绘和关联临床、病理和遗传实体,并了解潜在的疾病机制。在这篇综述中,我们总结了这一引人入胜的疾病的遗传、神经病理学和临床研究领域的快速发展现状。