Department of Pathology, Institute of Biomedicine, University of Gothenburg, Sahlgrenska University Hospital, 413 45 Gothenburg, Sweden.
Acta Neuropathol. 2013 Jan;125(1):3-18. doi: 10.1007/s00401-012-1024-2. Epub 2012 Aug 5.
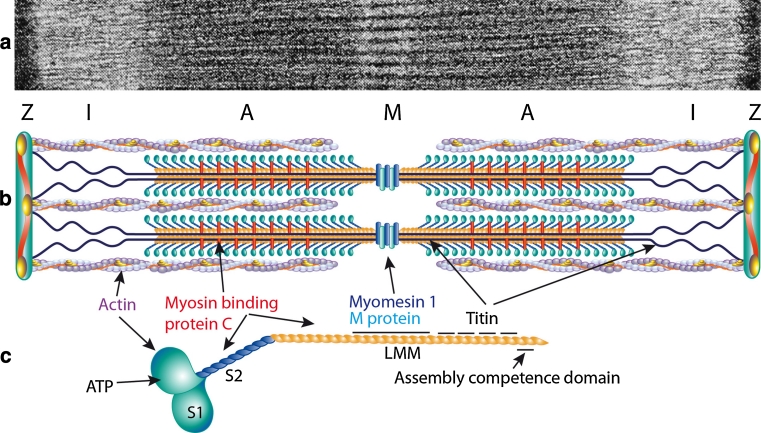
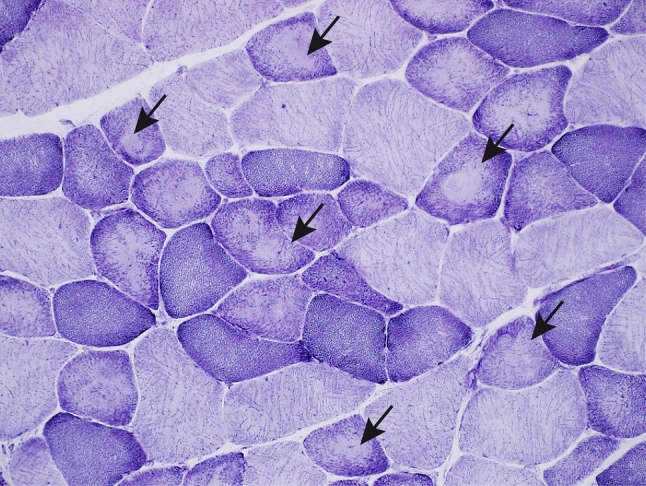
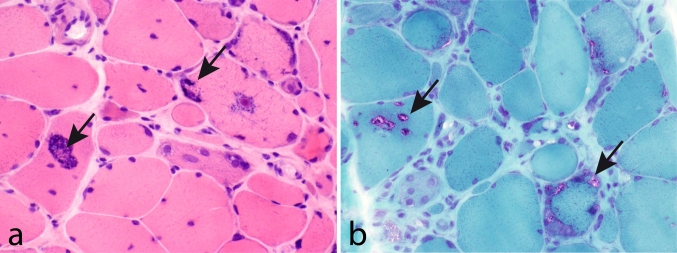
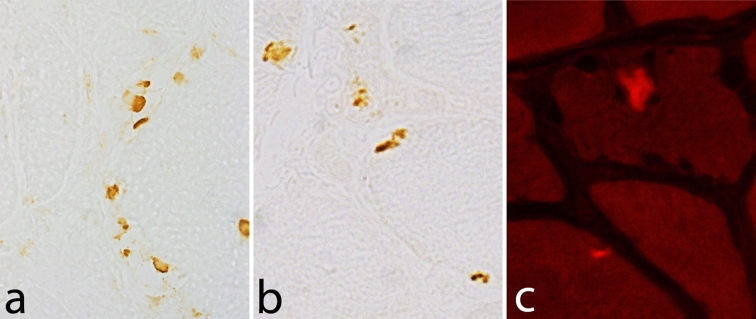
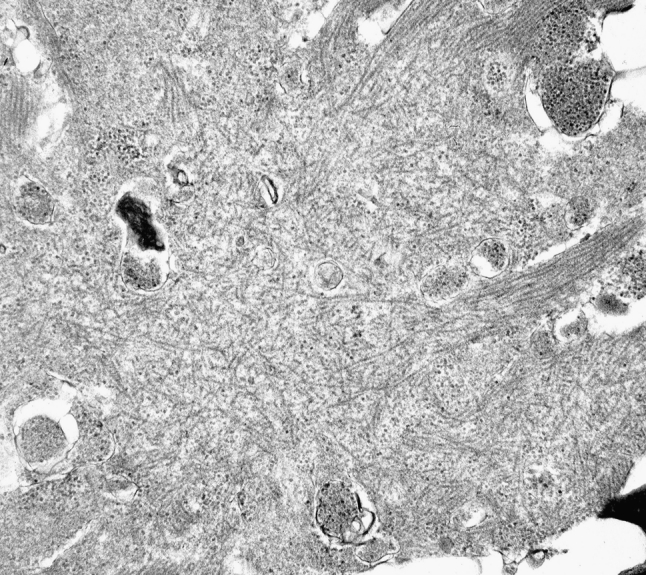
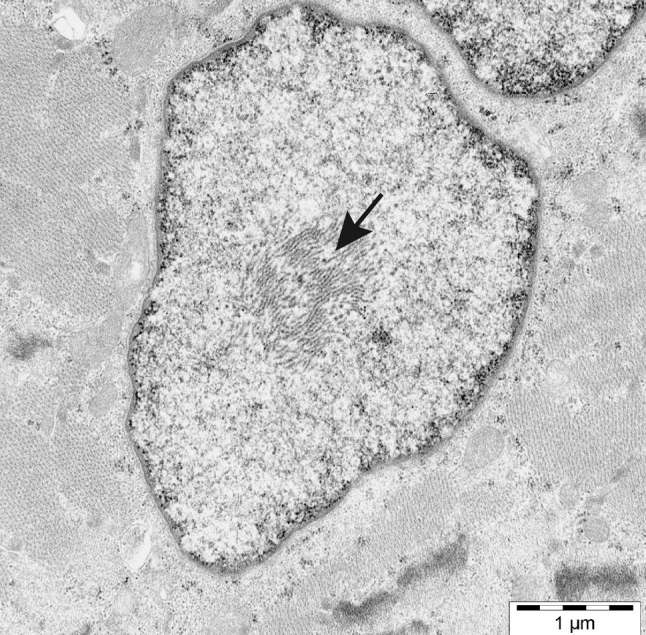
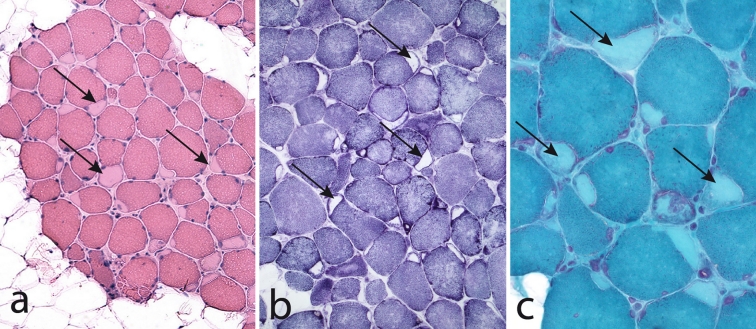
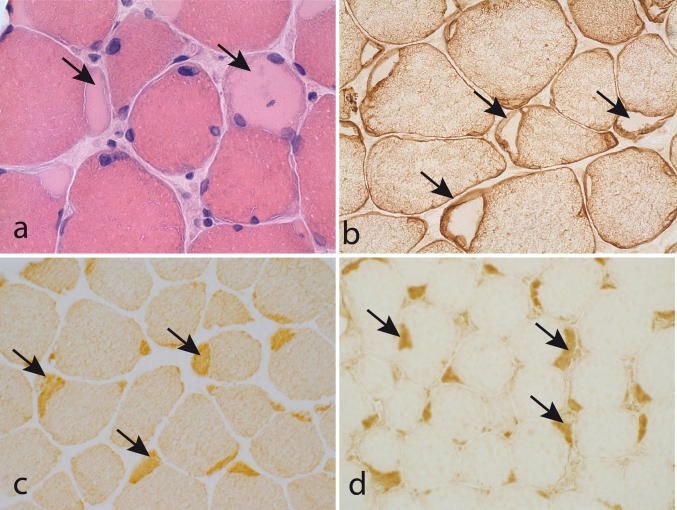
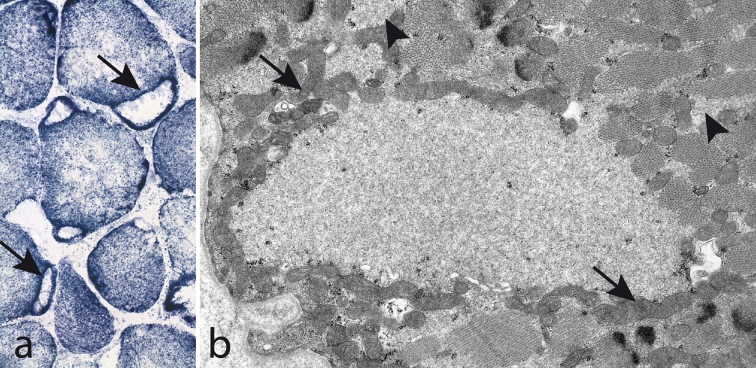
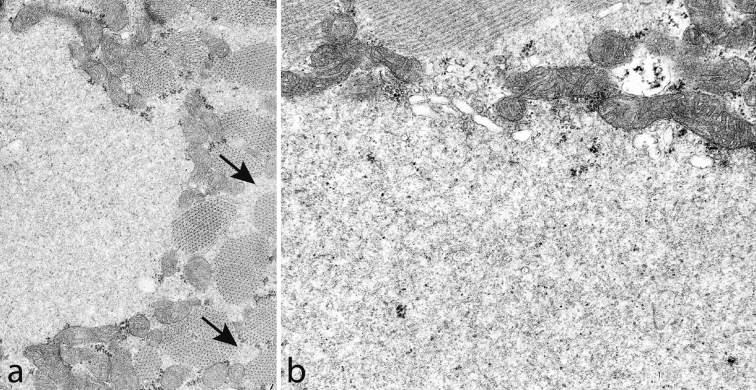
The myosin heavy chain (MyHC) is the molecular motor of muscle and forms the backbone of the sarcomere thick filaments. Different MyHC isoforms are of importance for the physiological properties of different muscle fiber types. Hereditary myosin myopathies have emerged as an important group of diseases with variable clinical and morphological expression depending on the mutated isoform and type and location of the mutation. Dominant mutations in developmental MyHC isoform genes (MYH3 and MYH8) are associated with distal arthrogryposis syndromes. Dominant or recessive mutations affecting the type IIa MyHC (MYH2) are associated with early-onset myopathies with variable muscle weakness and ophthalmoplegia as a consistent finding. Myopathies with scapuloperoneal, distal or limb-girdle muscle weakness including entities, such as myosin storage myopathy and Laing distal myopathy are the result of usually dominant mutations in the gene for slow/β cardiac MyHC (MYH7). Protein aggregation is part of the features in some of these myopathies. In myosin storage myopathy protein aggregates are formed by accumulation of myosin beneath the sarcolemma and between myofibrils. In vitro studies on the effects of different mutations associated with myosin storage myopathy and Laing distal myopathy indicate altered biochemical and biophysical properties of the light meromyosin, which is essential for thick filament assembly. Protein aggregates in the form of tubulofilamentous inclusions in association with vacuolated muscle fibers are present at late stage of dominant myosin IIa myopathy and sometimes in Laing distal myopathy. These protein aggregates exhibit features indicating defective degradation of misfolded proteins. In addition to protein aggregation and muscle fiber degeneration some of the myosin mutations cause functional impairment of the molecular motor adding to the pathogenesis of myosinopathies.
肌球蛋白重链(MyHC)是肌肉的分子马达,构成肌节粗丝的骨架。不同的 MyHC 同工型对于不同的肌纤维类型的生理特性很重要。遗传性肌球蛋白肌病已成为一组重要的疾病,其临床表现和形态学表现因突变同工型以及突变的类型和位置而异。发育性 MyHC 同工型基因(MYH3 和 MYH8)的显性突变与远端关节挛缩症综合征有关。影响 IIa 型 MyHC(MYH2)的显性或隐性突变与早发性肌病有关,其特征为不同程度的肌肉无力和眼肌麻痹。肩胛带、远端或肢体带肌无力的肌病,包括肌球蛋白贮积症和 Laing 远端肌病等,是由于慢/β 型心肌肌球蛋白(MYH7)基因的通常显性突变所致。蛋白聚集是其中一些肌病的特征之一。在肌球蛋白贮积症中,蛋白聚集体是由肌球蛋白在肌膜下和肌原纤维之间的积累形成的。对与肌球蛋白贮积症和 Laing 远端肌病相关的不同突变的体外研究表明,轻酶解肌球蛋白的生化和生物物理特性发生了改变,而轻酶解肌球蛋白对于粗丝组装是必不可少的。在显性肌球蛋白 IIa 肌病的晚期,有时在 Laing 远端肌病中,以管状纤维包涵体的形式存在蛋白聚集体。这些蛋白聚集体表现出表明错误折叠蛋白降解缺陷的特征。除了蛋白聚集和肌纤维退化外,一些肌球蛋白突变导致分子马达的功能障碍,这增加了肌球蛋白病的发病机制。