Zakham Fathiah, Aouane Othmane, Ussery David, Benjouad Abdelaziz, Ennaji Moulay Mustapha
Laboratoire de Virologie et Hygiène & Microbiologie, Faculté des Sciences et Techniques, BP 146, Mohammedia, 20650, Morocco.
Microb Inform Exp. 2012 Aug 28;2(1):7. doi: 10.1186/2042-5783-2-7.
The genus Mycobacterium comprises different species, among them the most contagious and infectious bacteria. The members of the complex Mycobacterium tuberculosis are the most virulent microorganisms that have killed human and other mammals since millennia. Additionally, with the many different mycobacterial sequences available, there is a crucial need for the visualization and the simplification of their data. In this present study, we aim to highlight a comparative genome, proteome and phylogeny analysis between twenty-one mycobacterial (Tuberculosis and non tuberculosis) strains using a set of computational and bioinformatics tools (Pan and Core genome plotting, BLAST matrix and phylogeny analysis).
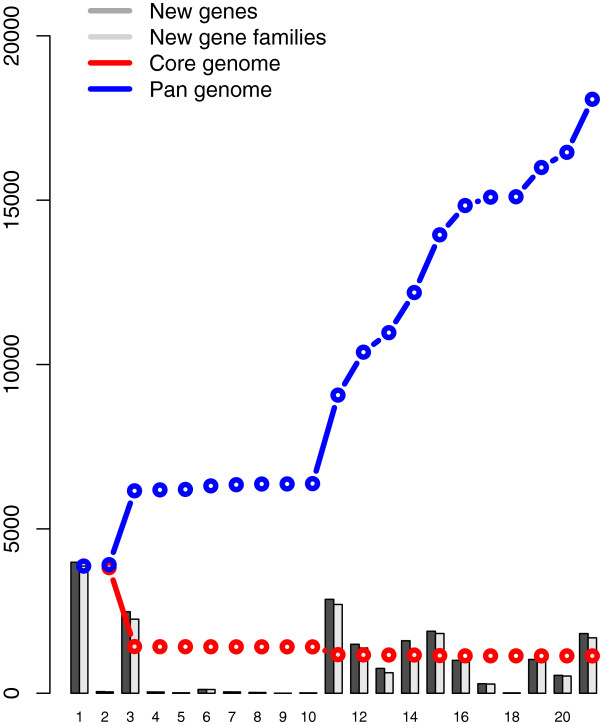
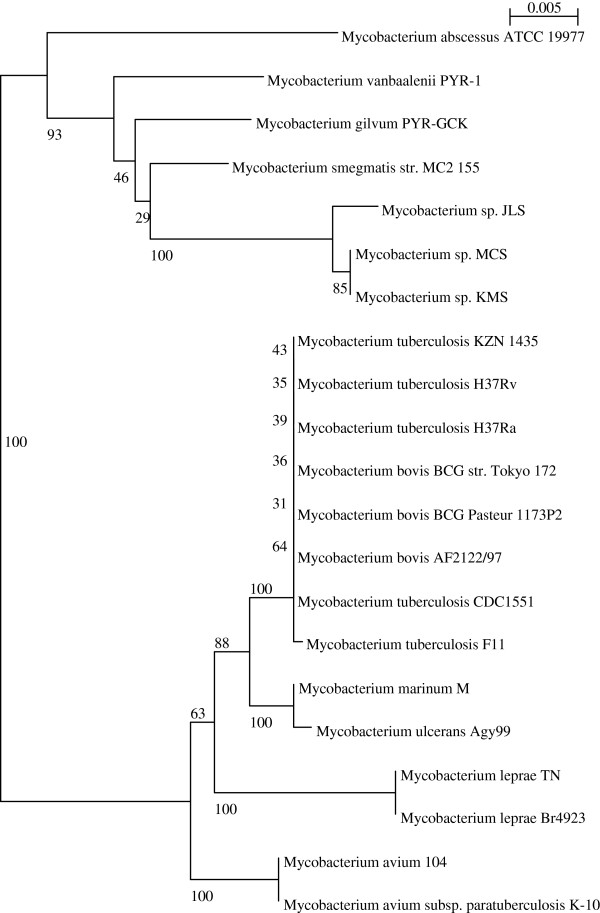
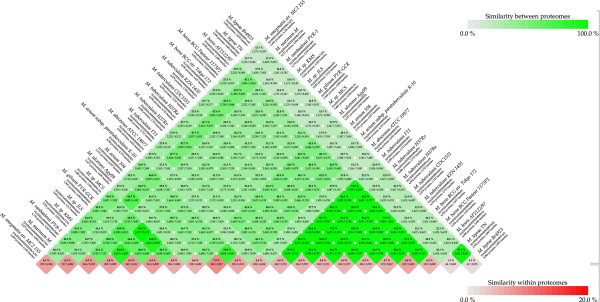
Considerably the result of pan and core genome Plotting demonstrated that less than 1250 Mycobacterium gene families are conserved across all species, and a total set of about 20,000 gene families within the Mycobacterium pan-genome of twenty one mycobacterial genomes.Viewing the BLAST matrix a high similarity was found among the species of the complex Mycobacterium tuberculosis and less conservation is found with other slow growing pathogenic mycobacteria.Phylogeny analysis based on both protein conservation, as well as rRNA clearly resolve known relationships between slow growing mycobacteria.
Mycobacteria include important pathogenic species for human and animals and the Mycobacterium tuberculosis complex is the most cause of death of the humankind. The comparative genome analysis could provide a new insight for better controlling and preventing these diseases.
分枝杆菌属包含不同的物种,其中包括传染性和感染性最强的细菌。结核分枝杆菌复合群的成员是数千年来导致人类和其他哺乳动物死亡的最具毒性的微生物。此外,由于有许多不同的分枝杆菌序列可用,因此迫切需要对其数据进行可视化和简化。在本研究中,我们旨在使用一组计算和生物信息学工具(泛基因组和核心基因组绘图、BLAST矩阵和系统发育分析),突出显示21种分枝杆菌(结核和非结核)菌株之间的比较基因组、蛋白质组和系统发育分析。
泛基因组和核心基因组绘图的结果表明,所有物种中保守的分枝杆菌基因家族不到1250个,在21个分枝杆菌基因组的分枝杆菌泛基因组中共有约20000个基因家族。查看BLAST矩阵发现,结核分枝杆菌复合群的物种之间具有高度相似性,而与其他生长缓慢的致病性分枝杆菌的保守性较低。基于蛋白质保守性以及rRNA的系统发育分析清楚地解析了生长缓慢的分枝杆菌之间的已知关系。
分枝杆菌包括对人类和动物重要的致病物种,结核分枝杆菌复合群是人类死亡的主要原因。比较基因组分析可为更好地控制和预防这些疾病提供新的见解。