Department of Computer Science, Stanford University, Stanford, California 94305, USA.
Genome Res. 2012 Sep;22(9):1735-47. doi: 10.1101/gr.136366.111.
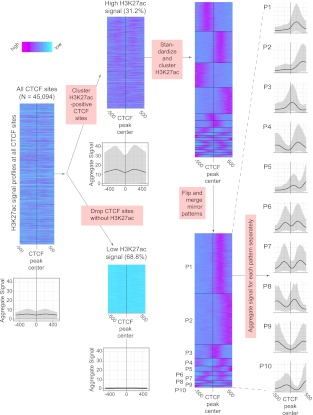
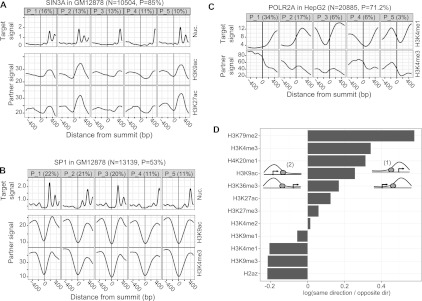
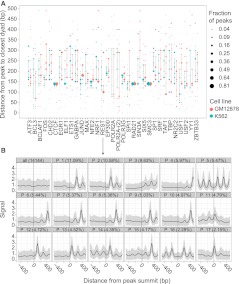
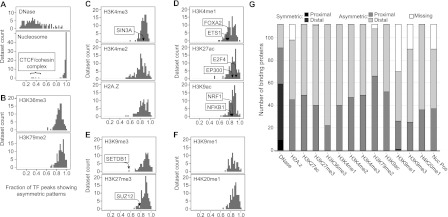
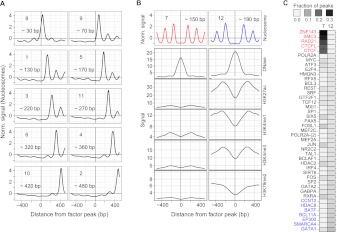
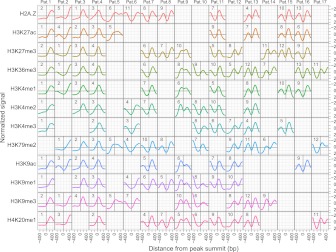
Gene regulation at functional elements (e.g., enhancers, promoters, insulators) is governed by an interplay of nucleosome remodeling, histone modifications, and transcription factor binding. To enhance our understanding of gene regulation, the ENCODE Consortium has generated a wealth of ChIP-seq data on DNA-binding proteins and histone modifications. We additionally generated nucleosome positioning data on two cell lines, K562 and GM12878, by MNase digestion and high-depth sequencing. Here we relate 14 chromatin signals (12 histone marks, DNase, and nucleosome positioning) to the binding sites of 119 DNA-binding proteins across a large number of cell lines. We developed a new method for unsupervised pattern discovery, the Clustered AGgregation Tool (CAGT), which accounts for the inherent heterogeneity in signal magnitude, shape, and implicit strand orientation of chromatin marks. We applied CAGT on a total of 5084 data set pairs to obtain an exhaustive catalog of high-resolution patterns of histone modifications and nucleosome positioning signals around bound transcription factors. Our analyses reveal extensive heterogeneity in how histone modifications are deposited, and how nucleosomes are positioned around binding sites. With the exception of the CTCF/cohesin complex, asymmetry of nucleosome positioning is predominant. Asymmetry of histone modifications is also widespread, for all types of chromatin marks examined, including promoter, enhancer, elongation, and repressive marks. The fine-resolution signal shapes discovered by CAGT unveiled novel correlation patterns between chromatin marks, nucleosome positioning, and sequence content. Meta-analyses of the signal profiles revealed a common vocabulary of chromatin signals shared across multiple cell lines and binding proteins.
功能元件(如增强子、启动子、绝缘子)的基因调控受到核小体重塑、组蛋白修饰和转录因子结合的相互作用的控制。为了增强我们对基因调控的理解,ENCODE 联盟生成了大量关于 DNA 结合蛋白和组蛋白修饰的 ChIP-seq 数据。我们还通过 MNase 消化和高深度测序生成了两条细胞系 K562 和 GM12878 的核小体定位数据。在这里,我们将 14 种染色质信号(12 种组蛋白标记物、DNase 和核小体定位)与 119 种 DNA 结合蛋白在大量细胞系中的结合位点相关联。我们开发了一种新的无监督模式发现方法,即聚类聚集工具(CAGT),该方法考虑了信号幅度、形状和染色质标记隐含链方向的固有异质性。我们总共对 5084 对数据集应用 CAGT,以获得围绕结合转录因子的组蛋白修饰和核小体定位信号的高分辨率模式的详尽目录。我们的分析揭示了组蛋白修饰沉积方式和核小体在结合位点周围定位方式的广泛异质性。除了 CTCF/黏合复合物外,核小体定位的不对称性占主导地位。核小体定位的不对称性也很普遍,所有类型的染色质标记物(包括启动子、增强子、延伸和抑制标记物)都存在这种现象。CAGT 发现的精细分辨率信号形状揭示了染色质标记物、核小体定位和序列内容之间的新的相关模式。信号图谱的元分析揭示了多个细胞系和结合蛋白共享的常见染色质信号词汇。