Ventura Ilenia, Russo Maria Teresa, De Nuccio Chiara, De Luca Gabriele, Degan Paolo, Bernardo Antonietta, Visentin Sergio, Minghetti Luisa, Bignami Margherita
Department of Environment and Primary Prevention, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Rome, Italy.
Department of Cell Biology and Neuroscience, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Rome, Italy.
Neurobiol Dis. 2013 Jan;49(12):148-58. doi: 10.1016/j.nbd.2012.09.002. Epub 2012 Sep 10.
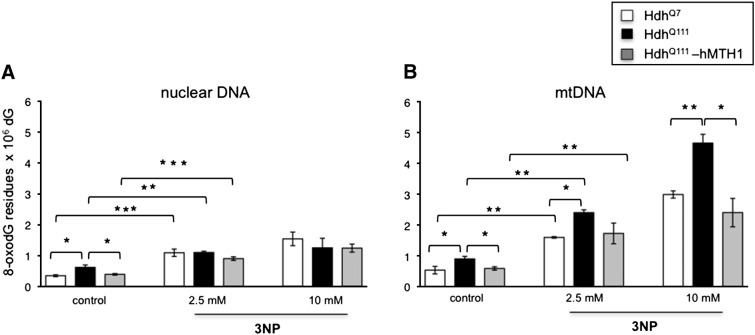
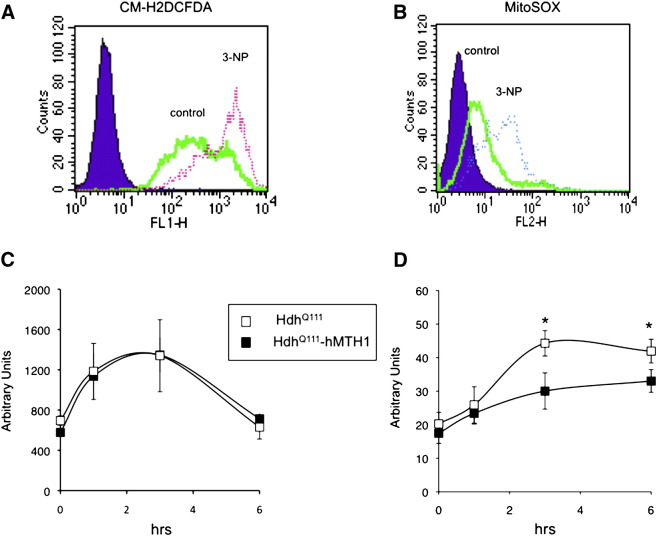
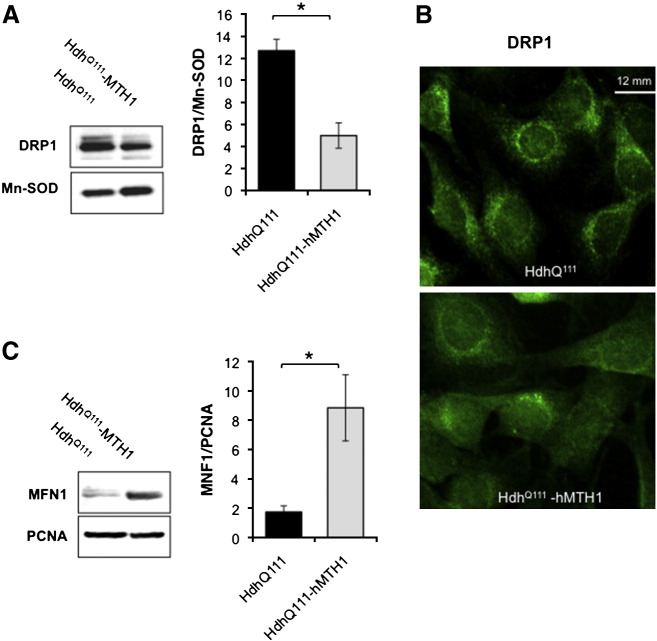
Huntington disease (HD) is a neurodegenerative disease caused by expansion of CAG repeats in the huntingtin (Htt) gene. The expression of hMTH1, the human hydrolase that degrades oxidized purine nucleoside triphosphates, grants protection in a chemical HD mouse model in which HD-like features are induced by the mitochondrial toxin 3-nitropropionic acid (3-NP). To further examine the relationship between oxidized dNTPs and HD-like neurodegeneration, we studied the effects of hMTH1 expression in a genetic cellular model for HD, such as striatal cells expressing mutant htt (Hdh(Q111)). hMTH1 expression protected these cells from 3-NP and H2O2-induced killing, by counteracting the mutant htt-dependent increased vulnerability and accumulation of nuclear and mitochondrial DNA 8-hydroxyguanine levels. hMTH1 expression reverted the decreased mitochondrial membrane potential characteristic of Hdh(Q111) cells and delayed the increase in mitochondrial reactive oxygen species associated with 3-NP treatment. Further indications of hMTH1-mediated mitochondrial protection are the partial reversion of 3-NP-induced alterations in mitochondrial morphology and the modulation of DRP1 and MFN1 proteins, which control fusion/fission rates of mitochondria. Finally, in line with the in vitro findings, upon 3-NP in vivo treatment, 8-hydroxyguanine levels in mitochondrial DNA from heart, muscle and brain are significantly lower in transgenic hMTH1-expressing mice than in wild-type animals.
亨廷顿舞蹈症(HD)是一种由亨廷顿蛋白(Htt)基因中CAG重复序列扩增引起的神经退行性疾病。人源水解酶hMTH1可降解氧化型嘌呤核苷三磷酸,在一种化学性HD小鼠模型中,hMTH1的表达具有保护作用,该模型中类似HD的特征由线粒体毒素3-硝基丙酸(3-NP)诱导产生。为了进一步研究氧化型脱氧核苷酸三磷酸(dNTPs)与类似HD的神经退行性变之间的关系,我们在HD的基因细胞模型中研究了hMTH1表达的影响,比如表达突变型htt(Hdh(Q111))的纹状体细胞。hMTH1的表达通过抵消突变型htt依赖性的易损性增加以及核DNA和线粒体DNA 8-羟基鸟嘌呤水平的积累,保护这些细胞免受3-NP和过氧化氢(H2O2)诱导的杀伤。hMTH1的表达恢复了Hdh(Q111)细胞特征性的线粒体膜电位降低,并延缓了与3-NP处理相关的线粒体活性氧增加。hMTH1介导的线粒体保护的进一步迹象包括3-NP诱导的线粒体形态改变的部分恢复以及对动力相关蛋白1(DRP1)和线粒体融合蛋白1(MFN1)的调节,这两种蛋白控制线粒体的融合/分裂速率。最后,与体外研究结果一致,在体内用3-NP处理后,转基因表达hMTH1的小鼠心脏、肌肉和大脑中线粒体DNA的8-羟基鸟嘌呤水平显著低于野生型动物。