Biomedicinal Information Research Center, National Institute of Advanced Industrial Science and Technology, Koto-ku, Tokyo, Japan.
Genome Biol Evol. 2012;4(11):1133-45. doi: 10.1093/gbe/evs075.
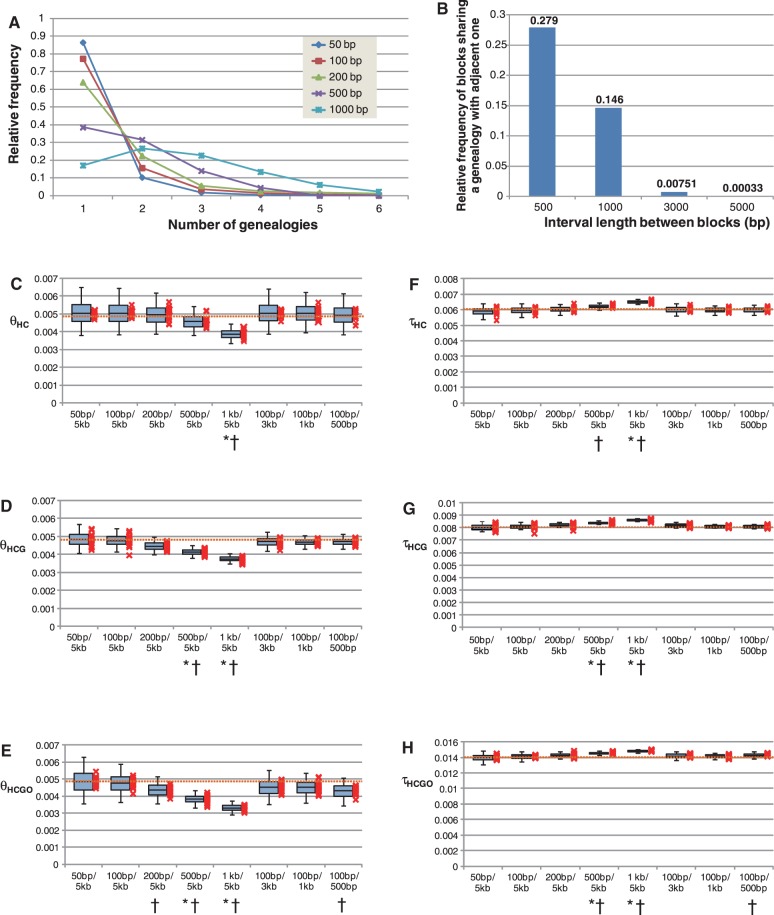
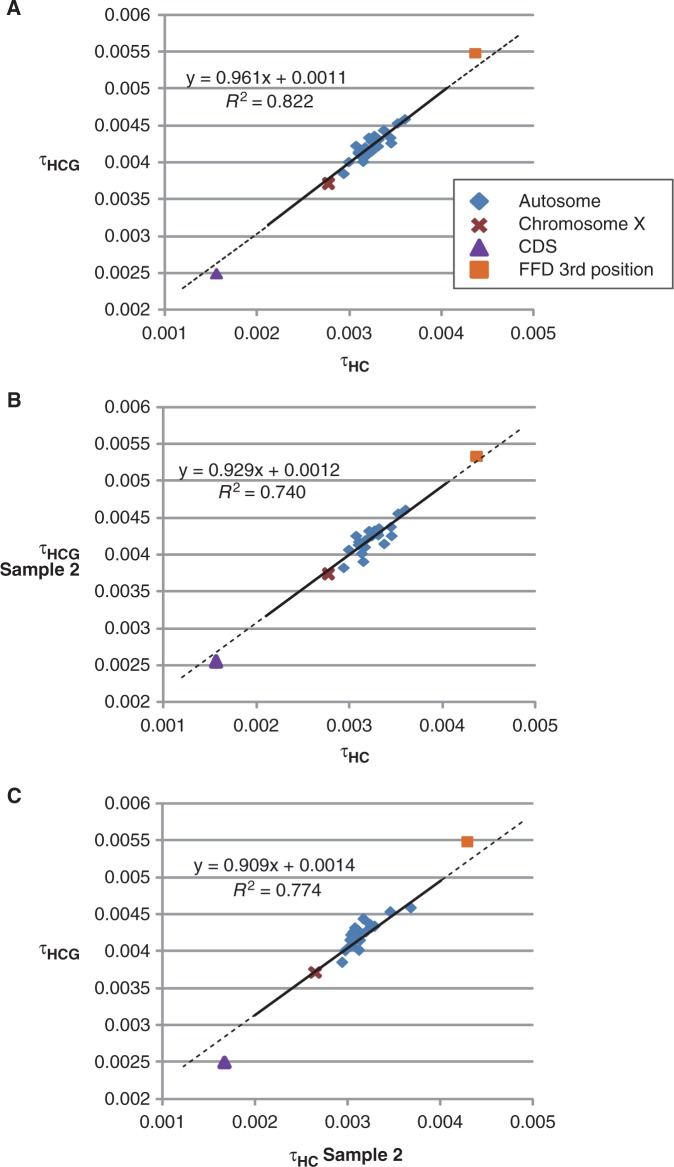
The demographic history of human would provide helpful information for identifying the evolutionary events that shaped the humanity but remains controversial even in the genomic era. To settle the controversies, we inferred the speciation times (T) and ancestral population sizes (N) in the lineage leading to human and great apes based on whole-genome alignment. A coalescence simulation determined the sizes of alignment blocks and intervals between them required to obtain recombination-free blocks with a high frequency. This simulation revealed that the size of the block strongly affects the parameter inference, indicating that recombination is an important factor for achieving optimum parameter inference. From the whole genome alignments (1.9 giga-bases) of human (H), chimpanzee (C), gorilla (G), and orangutan, 100-bp alignment blocks separated by ≥5-kb intervals were sampled and subjected to estimate τ = μT and θ = 4μgN using the Markov chain Monte Carlo method, where μ is the mutation rate and g is the generation time. Although the estimated τ(HC) differed across chromosomes, τ(HC) and τ(HCG) were strongly correlated across chromosomes, indicating that variation in τ is subject to variation in μ, rather than T, and thus, all chromosomes share a single speciation time. Subsequently, we estimated Ts of the human lineage from chimpanzee, gorilla, and orangutan to be 6.0-7.6, 7.6-9.7, and 15-19 Ma, respectively, assuming variable μ across lineages and chromosomes. These speciation times were consistent with the fossil records. We conclude that the speciation times in our recombination-free analysis would be conclusive and the speciation between human and chimpanzee was a single event.
人类的人口历史为识别塑造人类的进化事件提供了有用的信息,但即使在基因组时代,这仍然存在争议。为了解决这些争议,我们基于全基因组比对推断了人类和大猿类谱系的物种形成时间(T)和祖先种群大小(N)。 合并模拟确定了对齐块的大小和它们之间的间隔,以获得具有高频率的无重组块。该模拟表明块的大小强烈影响参数推断,表明重组是实现最佳参数推断的重要因素。从人类(H)、黑猩猩(C)、大猩猩(G)和猩猩(O)的全基因组比对(1.9 吉字节)中,选择了 100 个碱基对的对齐块,这些块由≥5-kb 间隔隔开,并采用马尔可夫链蒙特卡罗法估计 τ=μT 和 θ=4μgN,其中 μ 是突变率,g 是世代时间。虽然估计的 τ(HC)在染色体之间有所不同,但 τ(HC)和 τ(HCG)在染色体之间呈强相关,表明 τ 的变化受 μ 的变化而不是 T 的变化的影响,因此,所有染色体共享单一的物种形成时间。随后,我们假设不同谱系和染色体的 μ 不同,分别估计人类谱系从黑猩猩、大猩猩和猩猩的 Ts 为 6.0-7.6、7.6-9.7 和 15-19 Ma。这些物种形成时间与化石记录一致。我们得出结论,我们的无重组分析中的物种形成时间将是结论性的,人类和黑猩猩之间的物种形成是一个单一的事件。