Department of Computer Science, Strada Le Grazie 15, 37134 Verona, Italy.
BMC Genomics. 2012 Sep 17;13:485. doi: 10.1186/1471-2164-13-485.
In the post-genomic era several methods of computational genomics are emerging to understand how the whole information is structured within genomes. Literature of last five years accounts for several alignment-free methods, arisen as alternative metrics for dissimilarity of biological sequences. Among the others, recent approaches are based on empirical frequencies of DNA k-mers in whole genomes.
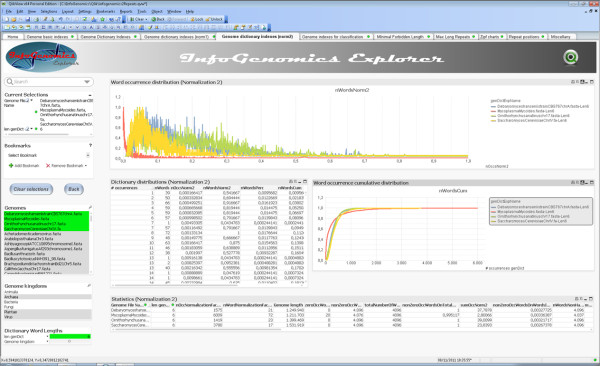
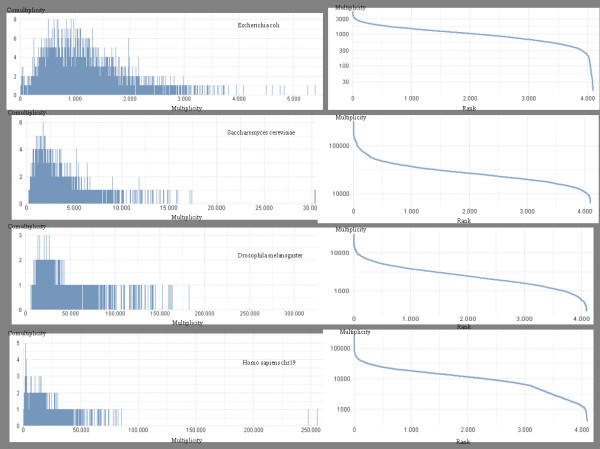
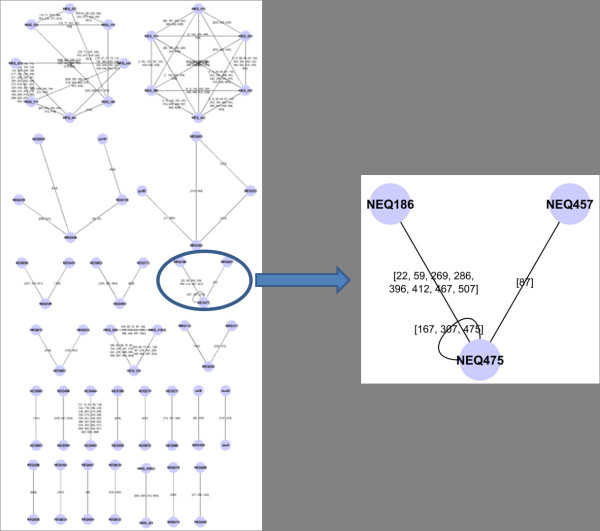
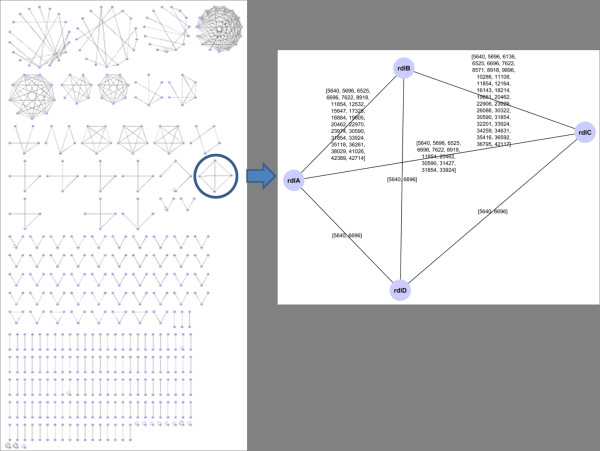
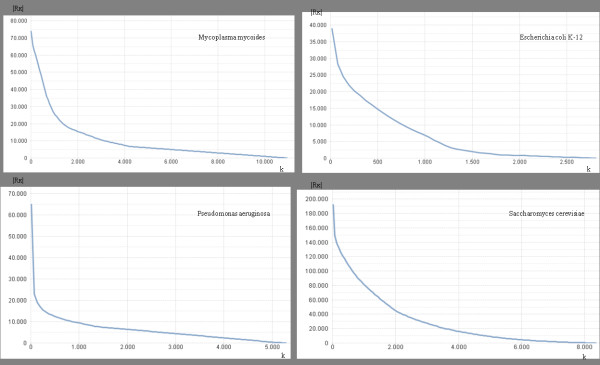
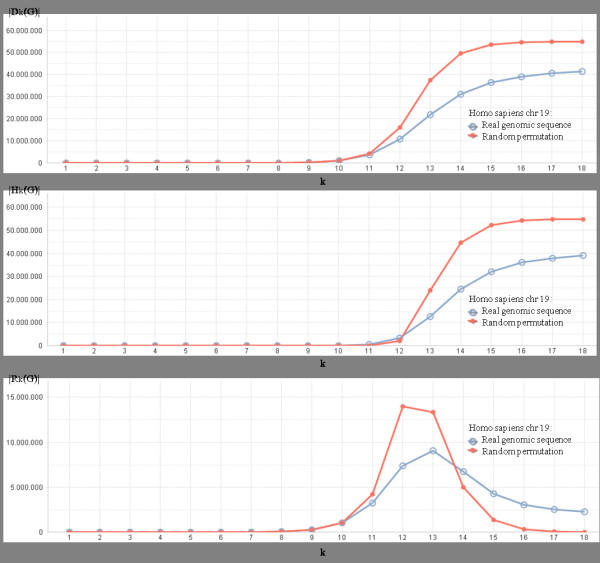
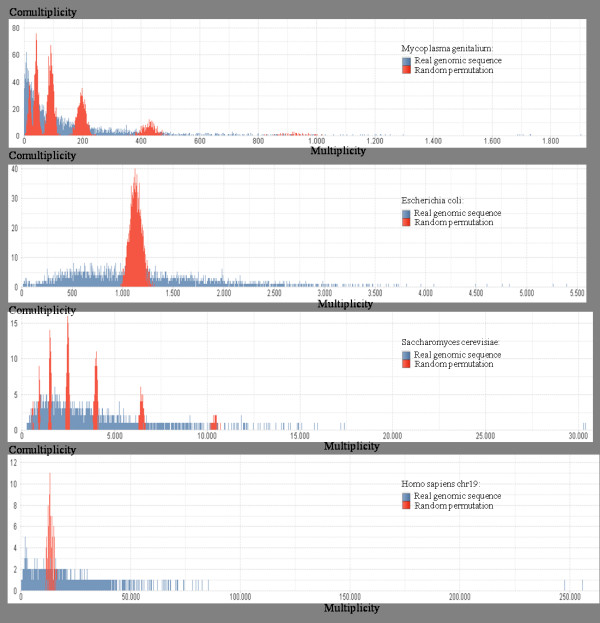
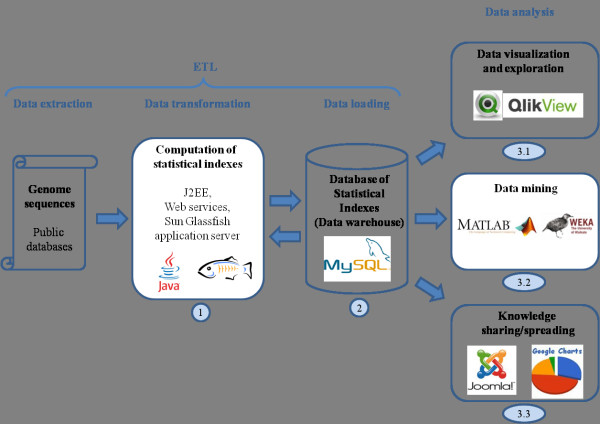
Any set of words (factors) occurring in a genome provides a genomic dictionary. About sixty genomes were analyzed by means of informational indexes based on genomic dictionaries, where a systemic view replaces a local sequence analysis. A software prototype applying a methodology here outlined carried out some computations on genomic data. We computed informational indexes, built the genomic dictionaries with different sizes, along with frequency distributions. The software performed three main tasks: computation of informational indexes, storage of these in a database, index analysis and visualization. The validation was done by investigating genomes of various organisms. A systematic analysis of genomic repeats of several lengths, which is of vivid interest in biology (for example to compute excessively represented functional sequences, such as promoters), was discussed, and suggested a method to define synthetic genetic networks.
We introduced a methodology based on dictionaries, and an efficient motif-finding software application for comparative genomics. This approach could be extended along many investigation lines, namely exported in other contexts of computational genomics, as a basis for discrimination of genomic pathologies.
在后基因组时代,出现了几种计算基因组学方法,以了解整个信息如何在基因组中构建。过去五年的文献记载了几种无比对方法,这些方法作为生物序列相似性的替代度量标准出现。在其他方法中,最近的方法基于整个基因组中 DNA k-mer 的经验频率。
基因组中出现的任何单词集(因子)都提供了基因组字典。通过基于基因组字典的信息指数对大约六十个基因组进行了分析,其中系统观点取代了局部序列分析。一个应用本文中概述的方法的软件原型对基因组数据进行了一些计算。我们计算了信息指数,构建了具有不同大小的基因组字典,以及频率分布。该软件执行了三个主要任务:计算信息指数、将这些指数存储在数据库中、指数分析和可视化。通过研究各种生物体的基因组来验证。讨论了对生物学非常感兴趣的各种长度的基因组重复的系统分析(例如,计算过度表示的功能序列,如启动子),并提出了一种定义合成遗传网络的方法。
我们介绍了一种基于字典的方法和一种用于比较基因组学的高效模体发现软件应用程序。这种方法可以沿着许多研究方向扩展,即在计算基因组学的其他背景下扩展,作为区分基因组病理学的基础。