Chen Chen, Zhao Jun-Fei, Huang Qiang, Wang Rui-Sheng, Zhang Xiang-Sun
Academy of Mathematics and Systems Science, Chinese Academy of Sciences, Beijing, PR China.
BMC Syst Biol. 2012;6 Suppl 1(Suppl 1):S7. doi: 10.1186/1752-0509-6-S1-S7. Epub 2012 Jul 16.
As protein domains are functional and structural units of proteins, a large proportion of protein-protein interactions (PPIs) are achieved by domain-domain interactions (DDIs), many computational efforts have been made to identify DDIs from experimental PPIs since high throughput technologies have produced a large number of PPIs for different species. These methods can be separated into two categories: deterministic and probabilistic. In deterministic methods, parsimony assumption has been utilized. Parsimony principle has been widely used in computational biology as the evolution of the nature is considered as a continuous optimization process. In the context of identifying DDIs, parsimony methods try to find a minimal set of DDIs that can explain the observed PPIs. This category of methods are promising since they can be formulated and solved easily. Besides, researches have shown that they can detect specific DDIs, which is often hard for many probabilistic methods. We notice that existing methods just view PPI networks as simply assembled by single interactions, but there is now ample evidence that PPI networks should be considered in a global (systematic) point of view for it exhibits general properties of complex networks, such as 'scale-free' and 'small-world'.
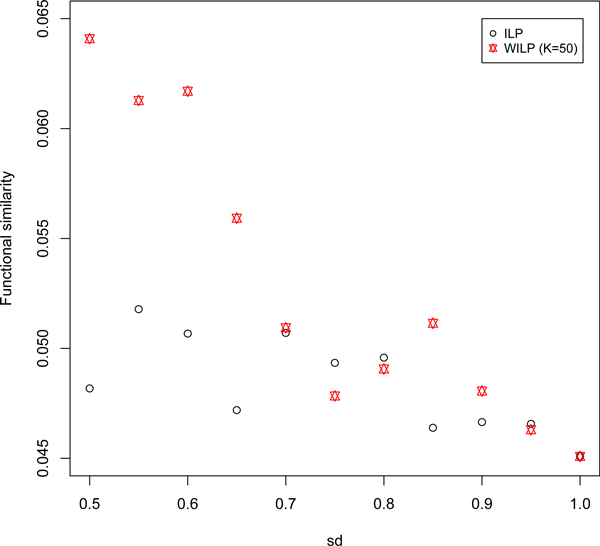
In this work, we integrate this global point of view into the parsimony-based model. Particularly, prior knowledge is extracted from these global properties by plausible reasoning and then taken as input. We investigate the role of the added information extensively through numerical experiments. Results show that the proposed method has improved performance, which confirms the biological meanings of the extracted prior knowledge.
This work provides us some clues for using these properties of complex networks in computational models and to some extent reveals the biological meanings underlying these general network properties.
由于蛋白质结构域是蛋白质的功能和结构单元,很大一部分蛋白质-蛋白质相互作用(PPI)是通过结构域-结构域相互作用(DDI)实现的。自从高通量技术为不同物种产生了大量PPI以来,人们已经进行了许多计算工作来从实验性PPI中识别DDI。这些方法可分为两类:确定性方法和概率性方法。在确定性方法中,采用了简约假设。简约原则在计算生物学中被广泛使用,因为自然界的进化被视为一个连续的优化过程。在识别DDI的背景下,简约方法试图找到一组最小的DDI,以解释观察到的PPI。这类方法很有前景,因为它们可以很容易地被公式化和求解。此外,研究表明它们可以检测到特定的DDI,而这对许多概率性方法来说往往很难。我们注意到,现有方法只是将PPI网络简单地视为由单个相互作用组装而成,但现在有充分的证据表明,应该从全局(系统)的角度来考虑PPI网络,因为它表现出复杂网络的一般特性,如“无标度”和“小世界”。
在这项工作中,我们将这种全局观点整合到基于简约的模型中。具体来说,通过合理推理从这些全局特性中提取先验知识,然后将其作为输入。我们通过数值实验广泛研究了添加信息的作用。结果表明,所提出的方法性能有所提高,这证实了所提取先验知识的生物学意义。
这项工作为我们在计算模型中利用复杂网络的这些特性提供了一些线索,并在一定程度上揭示了这些一般网络特性背后的生物学意义。