Yoon Dankyu, Kim Young Jin, Park Taesung
Interdisciplinary Program in Bioinformatics, Seoul National University, Seoul, 151-742, Korea.
BMC Syst Biol. 2012;6 Suppl 2(Suppl 2):S11. doi: 10.1186/1752-0509-6-S2-S11. Epub 2012 Dec 12.
A great success of the genome wide association study enabled us to give more attention on the personal genome and clinical application such as diagnosis and disease risk prediction. However, previous prediction studies using known disease associated loci have not been successful (Area Under Curve 0.55 ~ 0.68 for type 2 diabetes and coronary heart disease). There are several reasons for poor predictability such as small number of known disease-associated loci, simple analysis not considering complexity in phenotype, and a limited number of features used for prediction.
In this research, we investigated the effect of feature selection and prediction algorithm on the performance of prediction method thoroughly. In particular, we considered the following feature selection and prediction methods: regression analysis, regularized regression analysis, linear discriminant analysis, non-linear support vector machine, and random forest. For these methods, we studied the effects of feature selection and the number of features on prediction. Our investigation was based on the analysis of 8,842 Korean individuals genotyped by Affymetrix SNP array 5.0, for predicting smoking behaviors.
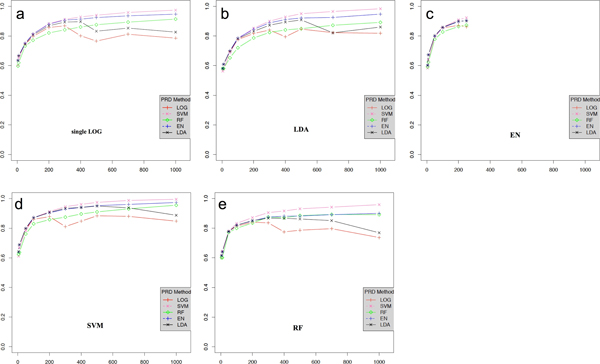
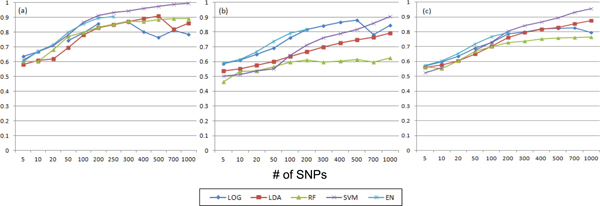
To observe the effect of feature selection methods on prediction performance, selected features were used for prediction and area under the curve score was measured. For feature selection, the performances of support vector machine (SVM) and elastic-net (EN) showed better results than those of linear discriminant analysis (LDA), random forest (RF) and simple logistic regression (LR) methods. For prediction, SVM showed the best performance based on area under the curve score. With less than 100 SNPs, EN was the best prediction method while SVM was the best if over 400 SNPs were used for the prediction.
Based on combination of feature selection and prediction methods, SVM showed the best performance in feature selection and prediction.
全基因组关联研究的巨大成功使我们能够更加关注个人基因组及其在诊断和疾病风险预测等临床应用中的作用。然而,以往使用已知疾病相关位点的预测研究并不成功(2型糖尿病和冠心病的曲线下面积为0.55至0.68)。预测能力较差有几个原因,比如已知疾病相关位点数量较少、未考虑表型复杂性的简单分析以及用于预测的特征数量有限。
在本研究中,我们全面研究了特征选择和预测算法对预测方法性能的影响。具体而言,我们考虑了以下特征选择和预测方法:回归分析、正则化回归分析、线性判别分析、非线性支持向量机和随机森林。对于这些方法,我们研究了特征选择和特征数量对预测的影响。我们的研究基于对8842名通过Affymetrix SNP阵列5.0进行基因分型的韩国个体的分析,以预测吸烟行为。
为了观察特征选择方法对预测性能的影响,将所选特征用于预测并测量曲线下面积得分。对于特征选择,支持向量机(SVM)和弹性网络(EN)的性能优于线性判别分析(LDA)、随机森林(RF)和简单逻辑回归(LR)方法。对于预测,基于曲线下面积得分,SVM表现最佳。当使用少于100个单核苷酸多态性(SNP)时,EN是最佳预测方法,而如果使用超过400个SNP进行预测,SVM则是最佳方法。
基于特征选择和预测方法的组合,SVM在特征选择和预测方面表现出最佳性能。