Department of Organismal and Evolutionary Biology, Harvard University, Cambridge, Massachusetts, USA.
PLoS Genet. 2012;8(12):e1003056. doi: 10.1371/journal.pgen.1003056. Epub 2012 Dec 20.
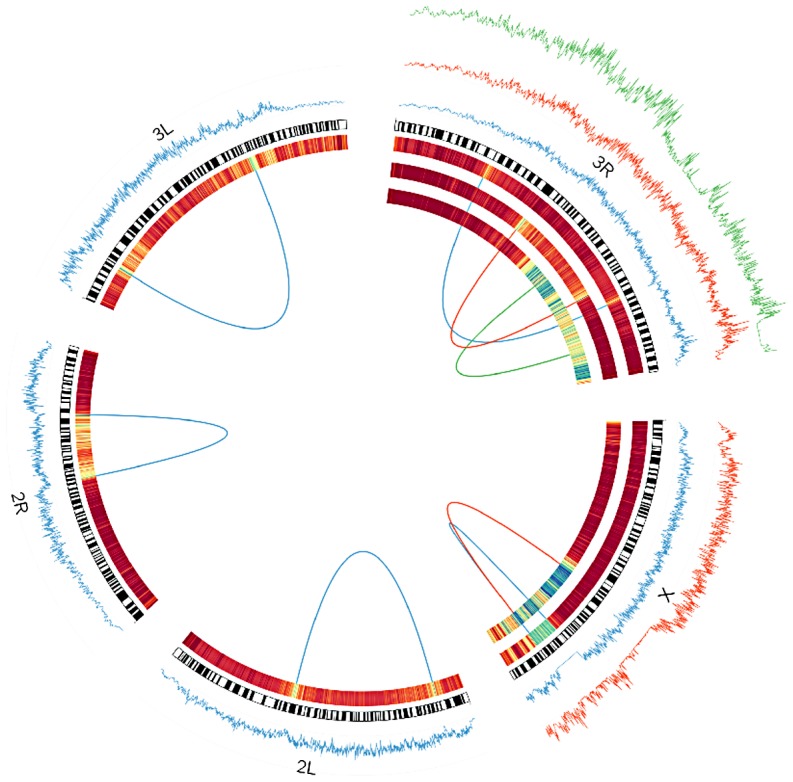
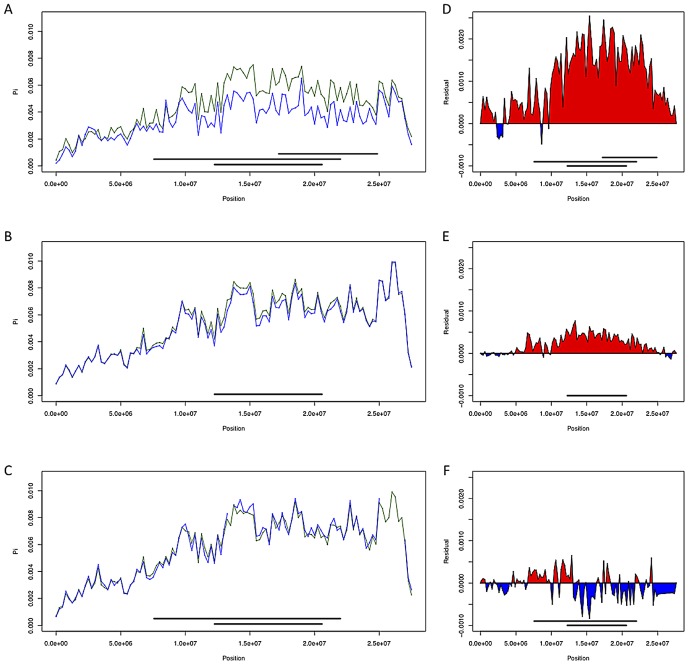
Chromosomal inversions have been an enduring interest of population geneticists since their discovery in Drosophila melanogaster. Numerous lines of evidence suggest powerful selective pressures govern the distributions of polymorphic inversions, and these observations have spurred the development of many explanatory models. However, due to a paucity of nucleotide data, little progress has been made towards investigating selective hypotheses or towards inferring the genealogical histories of inversions, which can inform models of inversion evolution and suggest selective mechanisms. Here, we utilize population genomic data to address persisting gaps in our knowledge of D. melanogaster's inversions. We develop a method, termed Reference-Assisted Reassembly, to assemble unbiased, highly accurate sequences near inversion breakpoints, which we use to estimate the age and the geographic origins of polymorphic inversions. We find that inversions are young, and most are African in origin, which is consistent with the demography of the species. The data suggest that inversions interact with polymorphism not only in breakpoint regions but also chromosome-wide. Inversions remain differentiated at low levels from standard haplotypes even in regions that are distant from breakpoints. Although genetic exchange appears fairly extensive, we identify numerous regions that are qualitatively consistent with selective hypotheses. Finally, we show that In(1)Be, which we estimate to be ∼60 years old (95% CI 5.9 to 372.8 years), has likely achieved high frequency via sex-ratio segregation distortion in males. With deeper sampling, it will be possible to build on our inferences of inversion histories to rigorously test selective models-particularly those that postulate that inversions achieve a selective advantage through the maintenance of co-adapted allele complexes.
自染色体倒位在黑腹果蝇中被发现以来,它们一直是群体遗传学家感兴趣的话题。大量的证据表明,强大的选择压力控制着多态性倒位的分布,这些观察结果激发了许多解释模型的发展。然而,由于核苷酸数据的缺乏,在研究选择假说或推断倒位的系统发育历史方面几乎没有取得进展,这些可以为倒位进化模型提供信息,并提出选择机制。在这里,我们利用群体基因组数据来解决我们对黑腹果蝇倒位知识的持续差距。我们开发了一种称为参考辅助组装的方法,用于组装在倒位断点附近无偏且高度准确的序列,我们使用该方法来估计多态性倒位的年龄和地理起源。我们发现,倒位很年轻,大多数起源于非洲,这与该物种的人口统计学一致。数据表明,倒位与多态性的相互作用不仅发生在断点区域,而且还发生在染色体范围内。即使在远离断点的区域,倒位与标准单倍型的分化程度仍然很低。尽管遗传交换似乎相当广泛,但我们确定了许多区域与选择假说定性一致。最后,我们表明,我们估计年龄为 60 岁左右的 In(1)Be(95%置信区间为 5.9 至 372.8 岁)可能通过雄性的性别比例分离偏度而达到高频。通过更深入的采样,有可能在我们对倒位历史的推断的基础上,严格检验选择模型,特别是那些假设倒位通过维持共适应等位基因复合物获得选择性优势的模型。