Department of Pharmacology, University of Oxford, Oxford, United Kingdom.
PLoS One. 2012;7(12):e52790. doi: 10.1371/journal.pone.0052790. Epub 2012 Dec 28.
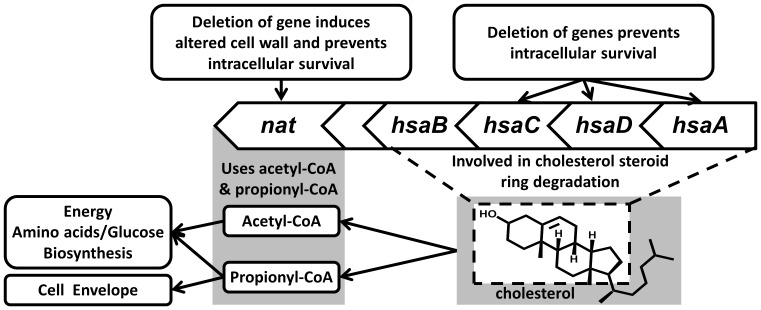
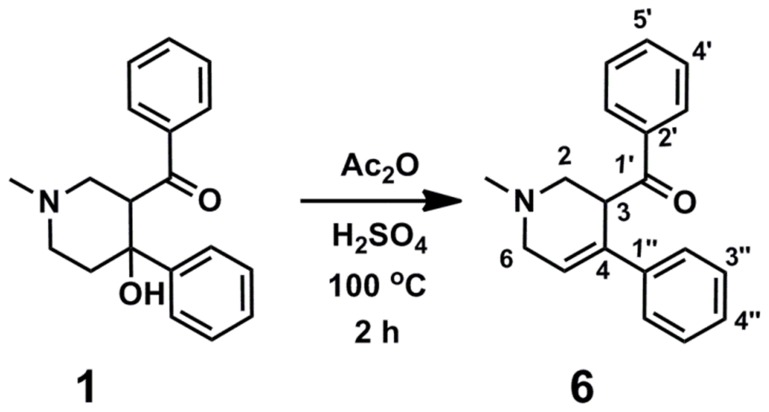
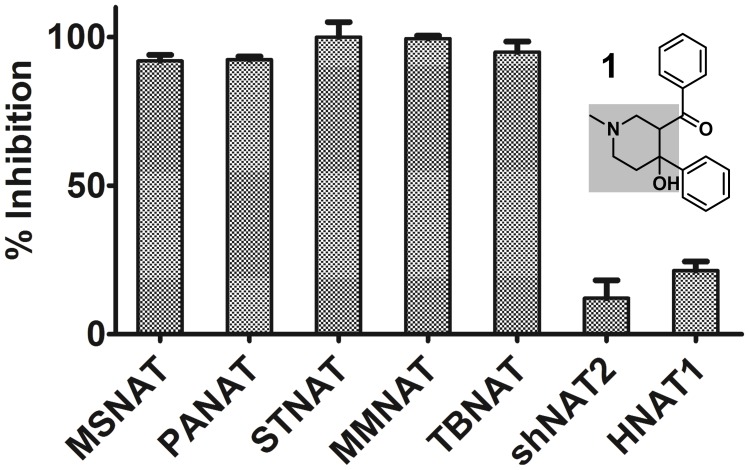
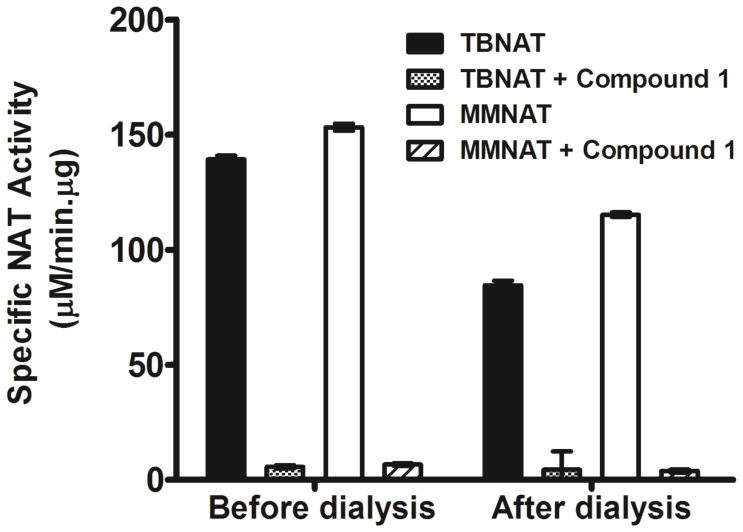
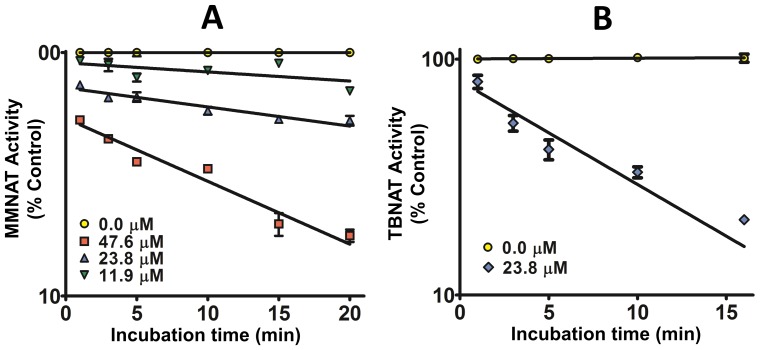
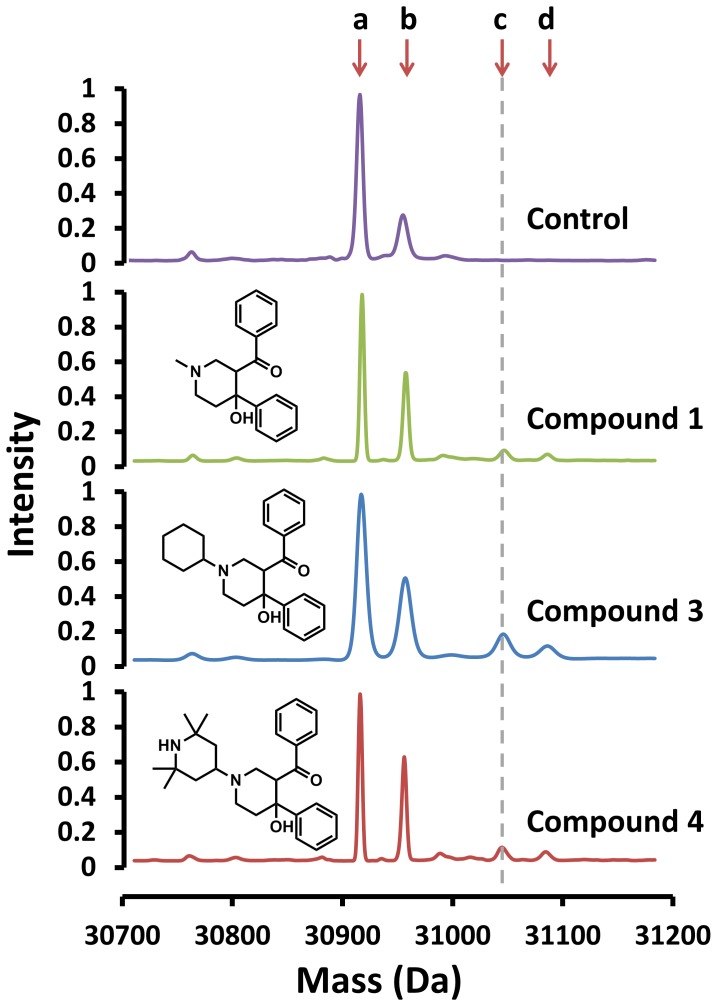
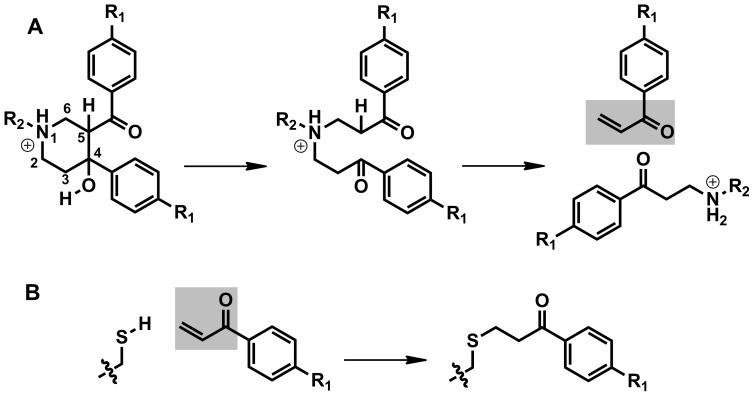
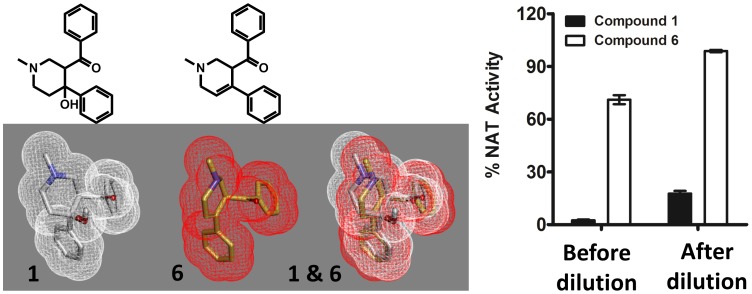
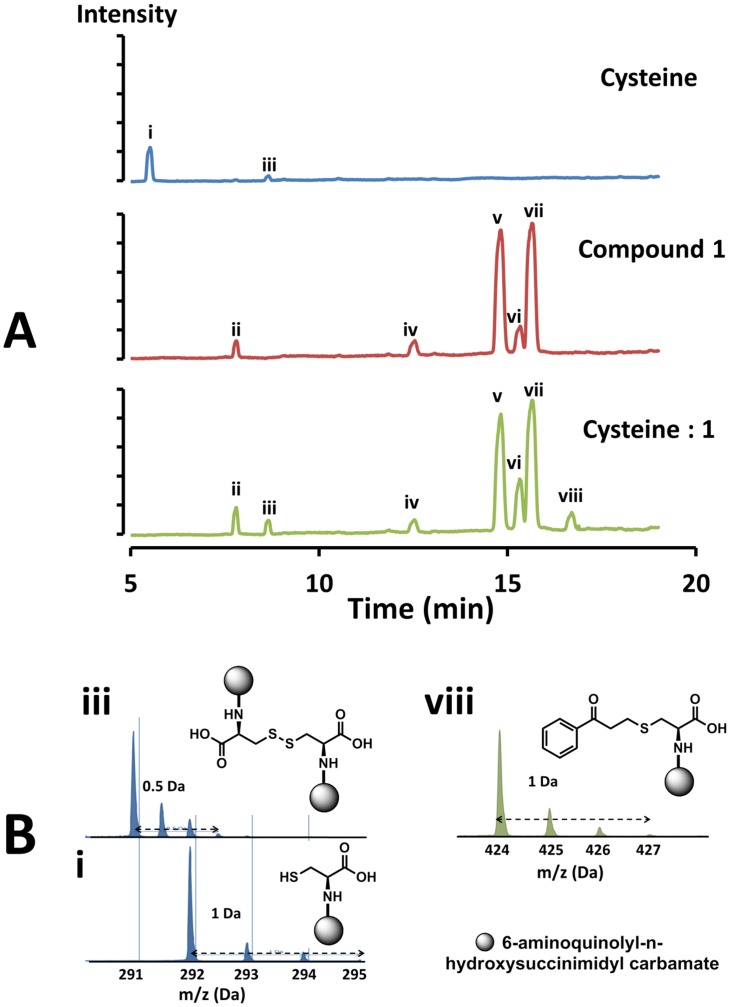
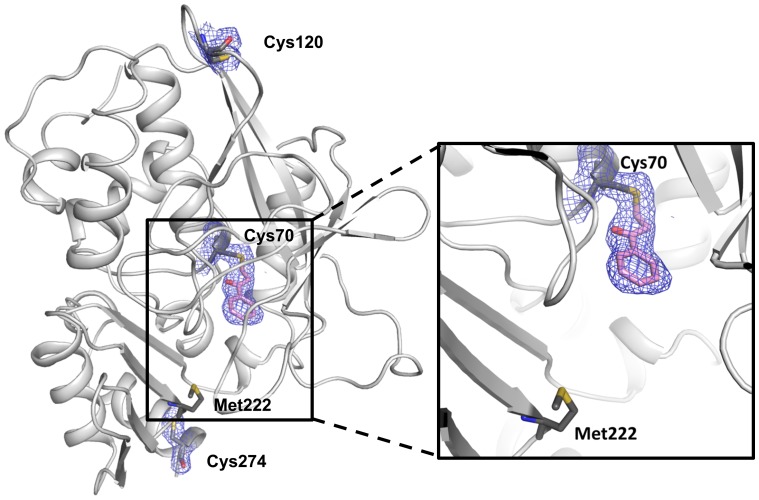
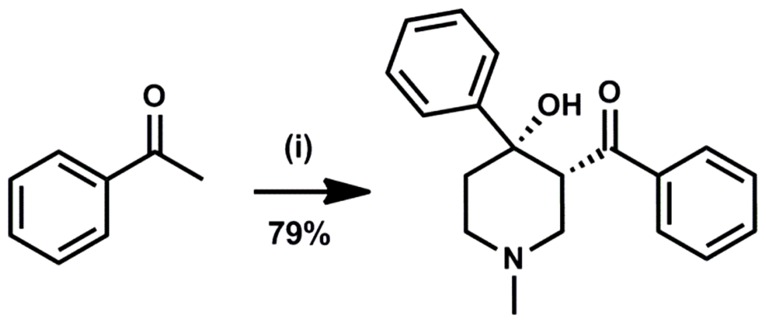
Latent M. tuberculosis infection presents one of the major obstacles in the global eradication of tuberculosis (TB). Cholesterol plays a critical role in the persistence of M. tuberculosis within the macrophage during latent infection. Catabolism of cholesterol contributes to the pool of propionyl-CoA, a precursor that is incorporated into cell-wall lipids. Arylamine N-acetyltransferase (NAT) is encoded within a gene cluster that is involved in the cholesterol sterol-ring degradation and is essential for intracellular survival. The ability of the NAT from M. tuberculosis (TBNAT) to utilise propionyl-CoA links it to the cholesterol-catabolism pathway. Deleting the nat gene or inhibiting the NAT enzyme prevents intracellular survival and results in depletion of cell-wall lipids. TBNAT has been investigated as a potential target for TB therapies. From a previous high-throughput screen, 3-benzoyl-4-phenyl-1-methylpiperidinol was identified as a selective inhibitor of prokaryotic NAT that exhibited antimycobacterial activity. The compound resulted in time-dependent irreversible inhibition of the NAT activity when tested against NAT from M. marinum (MMNAT). To further evaluate the antimycobacterial activity and the NAT inhibition of this compound, four piperidinol analogues were tested. All five compounds exert potent antimycobacterial activity against M. tuberculosis with MIC values of 2.3-16.9 µM. Treatment of the MMNAT enzyme with this set of inhibitors resulted in an irreversible time-dependent inhibition of NAT activity. Here we investigate the mechanism of NAT inhibition by studying protein-ligand interactions using mass spectrometry in combination with enzyme analysis and structure determination. We propose a covalent mechanism of NAT inhibition that involves the formation of a reactive intermediate and selective cysteine residue modification. These piperidinols present a unique class of antimycobacterial compounds that have a novel mode of action different from known anti-tubercular drugs.
潜伏性结核分枝杆菌感染是全球结核病(TB)根除的主要障碍之一。胆固醇在潜伏感染期间分枝杆菌在巨噬细胞内的持续存在中起着关键作用。胆固醇的分解代谢有助于丙酸酰辅酶 A 的池的形成,丙酸酰辅酶 A 是一种前体,被纳入细胞壁脂质中。芳基胺 N-乙酰转移酶(NAT)编码在一个基因簇中,该基因簇参与胆固醇甾环降解,是细胞内生存所必需的。分枝杆菌(TBNAT)的 NAT 利用丙酸酰辅酶 A 的能力将其与胆固醇分解代谢途径联系起来。删除 nat 基因或抑制 NAT 酶可防止细胞内生存并导致细胞壁脂质耗尽。TBNAT 已被研究作为结核病治疗的潜在靶点。从之前的高通量筛选中,鉴定出 3-苯甲酰-4-苯基-1-甲基哌啶醇是一种选择性抑制原核 NAT 的抑制剂,具有抗分枝杆菌活性。当针对 M. marinum(MMNAT)的 NAT 进行测试时,该化合物导致 NAT 活性的时间依赖性不可逆抑制。为了进一步评估该化合物的抗分枝杆菌活性和 NAT 抑制作用,测试了四种哌啶醇类似物。所有五种化合物均对结核分枝杆菌具有很强的抗分枝杆菌活性,MIC 值为 2.3-16.9µM。用这组抑制剂处理 MMNAT 酶会导致 NAT 活性的不可逆时间依赖性抑制。在这里,我们通过使用质谱结合酶分析和结构测定来研究蛋白质-配体相互作用,研究 NAT 抑制的机制。我们提出了 NAT 抑制的共价机制,涉及形成反应性中间产物和选择性半胱氨酸残基修饰。这些哌啶醇类化合物代表了一类具有独特抗分枝杆菌化合物,其作用模式与已知的抗结核药物不同。