Center for non-coding RNA in Technology and Health, University of Copenhagen, Frederiksberg, Denmark.
Hum Mutat. 2013 Apr;34(4):546-56. doi: 10.1002/humu.22273.
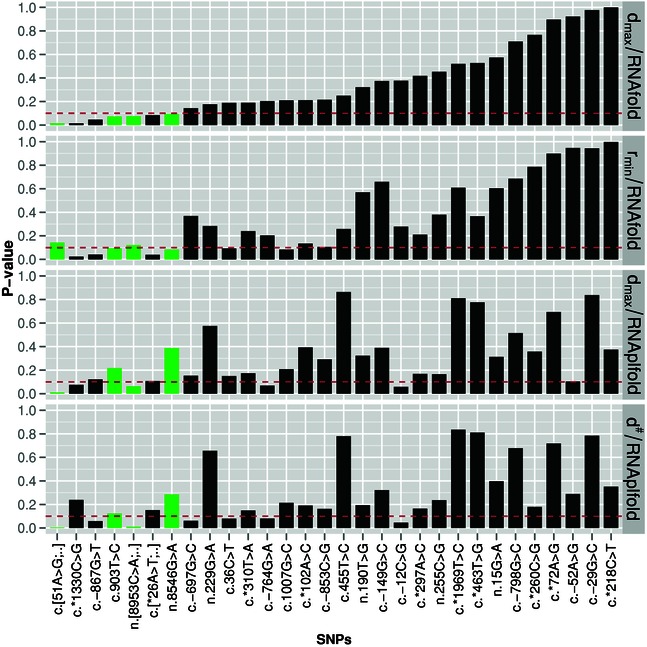
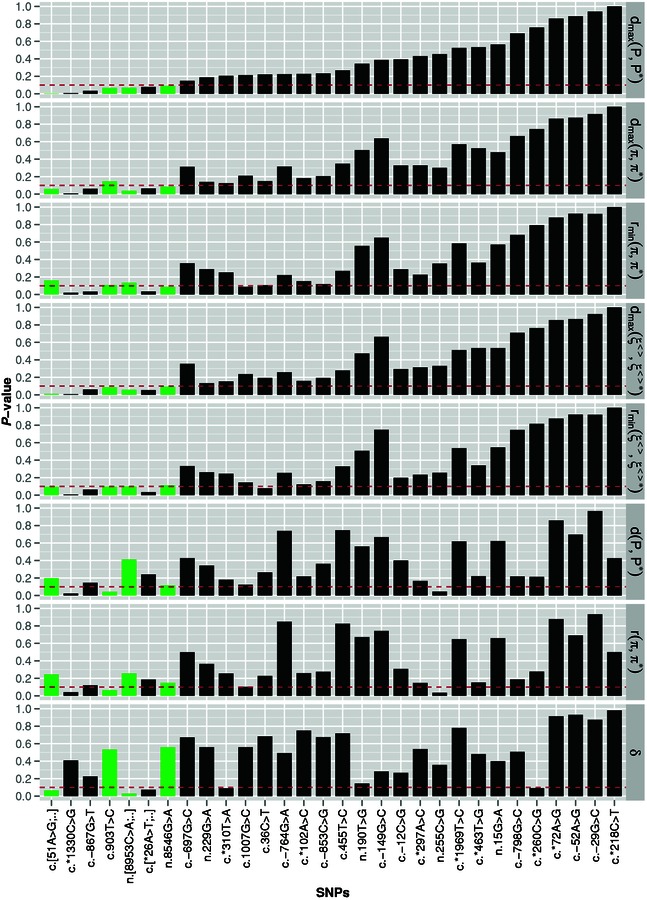
Structural characteristics are essential for the functioning of many noncoding RNAs and cis-regulatory elements of mRNAs. SNPs may disrupt these structures, interfere with their molecular function, and hence cause a phenotypic effect. RNA folding algorithms can provide detailed insights into structural effects of SNPs. The global measures employed so far suffer from limited accuracy of folding programs on large RNAs and are computationally too demanding for genome-wide applications. Here, we present a strategy that focuses on the local regions of maximal structural change between mutant and wild-type. These local regions are approximated in a "screening mode" that is intended for genome-wide applications. Furthermore, localized regions are identified as those with maximal discrepancy. The mutation effects are quantified in terms of empirical P values. To this end, the RNAsnp software uses extensive precomputed tables of the distribution of SNP effects as function of length and GC content. RNAsnp thus achieves both a noise reduction and speed-up of several orders of magnitude over shuffling-based approaches. On a data set comprising 501 SNPs associated with human-inherited diseases, we predict 54 to have significant local structural effect in the untranslated region of mRNAs.
结构特征对于许多非编码 RNA 和 mRNA 的顺式调控元件的功能至关重要。SNP 可能破坏这些结构,干扰其分子功能,从而导致表型效应。RNA 折叠算法可以提供 SNP 结构影响的详细信息。迄今为止,所采用的全局度量标准在大型 RNA 上的折叠程序准确性有限,并且对于全基因组应用来说计算要求过高。在这里,我们提出了一种策略,该策略侧重于突变型和野生型之间最大结构变化的局部区域。这些局部区域在“筛选模式”中进行近似处理,该模式适用于全基因组应用。此外,确定了具有最大差异的局部区域。突变效应以经验 P 值来量化。为此,RNAsnp 软件使用了广泛的预先计算的 SNP 效应分布表,这些表是作为长度和 GC 含量的函数。因此,与基于随机打乱的方法相比,RNAsnp 在减少噪声和提高速度方面提高了几个数量级。在包含 501 个与人类遗传疾病相关的 SNP 的数据集上,我们预测 54 个 SNP 在 mRNA 的非翻译区具有显著的局部结构效应。